Genome Editing of Human Primary T Cells Using CRISPR-Cas9
- Document # 27155
- Version 2.0.2
- Jun 2023
Introduction
Through targeted modification of specific genes or regulatory regions with CRISPR genome-editing technology, researchers can now rapidly generate precise genetic models to study normal and diseased cell physiology. Beyond genetic manipulation for research purposes, CRISPR-Cas9 genome editing also holds great potential for therapeutic applications, including immunotherapy and regenerative medicine. Early attempts to apply CRISPR-Cas9 for genome editing in human primary T cells used either viral vectors1,2 or plasmids3,4 for Cas9 and gRNA expression, resulting in low targeting efficiency and high toxicity. More recently, ribonucleoprotein (RNP)-based CRISPR-Cas9 expression systems have achieved high efficacy across a number of targets.5-7
The following document provides instructions for genome editing of human primary T cells using an RNP-based CRISPR-Cas9 system, including optimization of pre- and post-editing culture conditions and methods to evaluate genome editing efficiency. The protocol details instructions for isolation and activation of primary human T cells, preparation of a CRISPR-Cas9 RNP complex using sgRNA or crRNA:tracrRNA duplexes, and delivery of RNP complex into activated primary T cells using electroporation (Figure 1). We describe one strategy to generate knockout primary T cells with high efficiency and present a case study and data on CRISPR-Cas9 genome editing of the TCRαβ locus across a number of activation conditions; see the discussion section of this document for alternative strategies and expected outcomes, as well as protocol modifications.
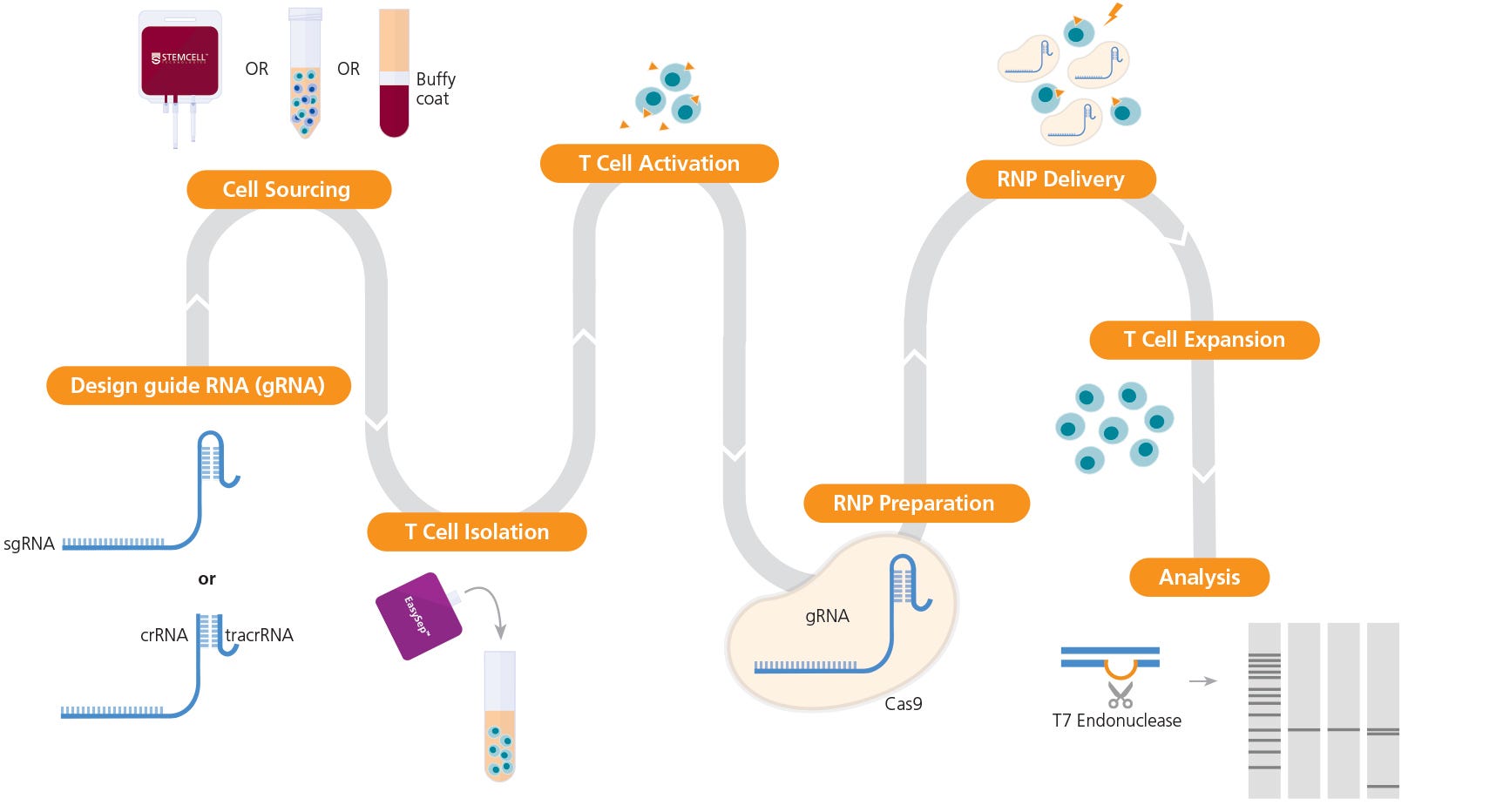
Figure 1. Experimental Workflow for T Cell Genome Editing
The ArciTect™ sgRNA (single guide RNA) or ArciTect™ crRNA (CRISPR RNA) sequences can be designed using the CRISPR Design Tool once a target locus for editing is identified. Human primary T cells* can be sourced from STEMCELL or isolated from a number of sources, including whole blood, buffy coat, washed leukapheresis samples, or peripheral blood mononuclear cells (PBMCs), using STEMCELL column-free cell separation technology, including EasySep™. Next, T cells are activated with either ImmunoCult™ CD3/CD28 T Cell Activator or ImmunoCult™ CD3/CD28/CD2 T Cell Activator. The ArciTect™ CRISPR-Cas9 RNP is then prepared and delivered into T cells using electroporation, and cells are expanded in ImmunoCult™-XF T Cell Expansion Medium. Editing efficiency can be analyzed using the ArciTect™ T7 Endonuclease I Kit or flow cytometry, if the experimental design permits. Orange triangles indicate ImmunoCult™ Human T Cell Activator.
*Certain products are only available in select territories. Please contact your local Sales representative or the Product & Scientific Support team at techsupport@stemcell.com for further information.
Background
The mechanism of CRISPR-Cas9 genome editing is shown in Figure 1. The CRISPR-Cas9 system is composed of two key components: a guide RNA (gRNA) and a CRISPR-associated endonuclease (Cas) protein. Briefly, Cas9 endonuclease is targeted to the desired genomic locus through complementary base pairing of the associated single guide RNA (sgRNA) or CRISPR RNA (crRNA) at a site 5’ to a protospacer adjacent motif (PAM) sequence (NGG for Streptococcus pyogenes Cas9). The crRNA partners with a trans-acting CRISPR RNA (tracrRNA), which acts as a scaffold for the interaction with Cas9, to form the guide RNA (gRNA). Once complexed with the gRNA, Cas9 is targeted to the desired genomic locus and will generate a DNA break 3 to 4 bases upstream of the PAM sequence. Endogenous DNA repair systems then function to repair the DNA break. Non-homologous end joining (NHEJ) mediates direct ligation of the broken DNA molecule, and due to the error-prone nature of NHEJ, this can result in the formation of insertion or deletion (INDEL) mutations. If a donor DNA sequence template is provided, knock-in of precise DNA sequences can be accomplished through the homology-directed repair (HDR) pathway (Figure 2).
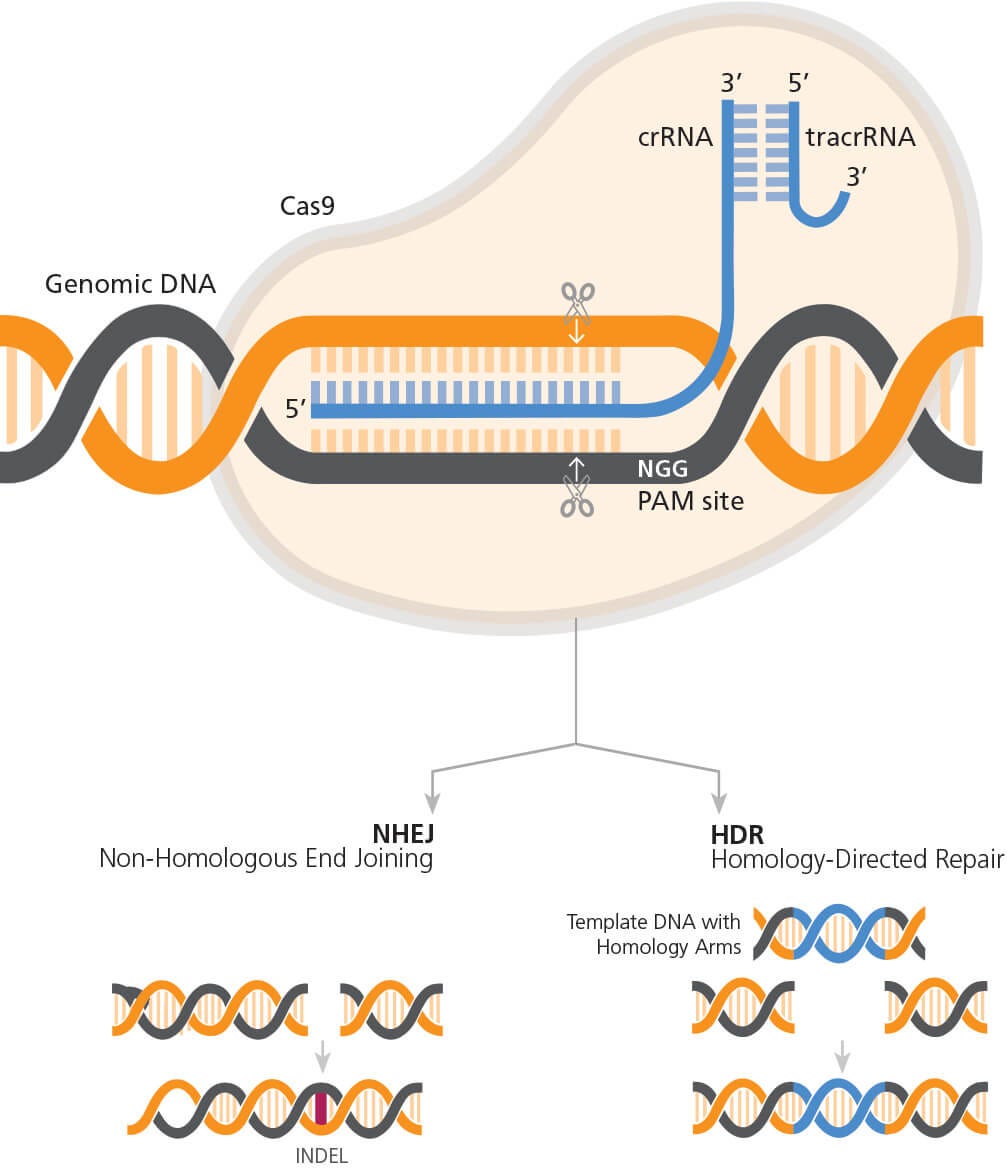
Figure 2. General Mechanism of CRISPR-Cas9 Genome Editing
The RNP complex (Cas9 partnered with gRNA) orientation with respect to target and PAM sites. Cas9 generates a DNA break 3 - 4 bp upstream of the PAM sequence, triggering endogenous DNA repair pathways. This can result in the formation of INDELs if repair occurs via the NHEJ pathway or knock-in of precise sequence changes via the HDR pathway, if template DNA is included in experimental design.
While this technology has been successfully applied in numerous cell lines, its application in primary human immune cells has been hampered by challenges in efficient delivery and expression of CRISPR-Cas9 components. As mentioned before, early attempts to apply CRISPR-Cas9 for genome editing in primary human T cells used either viral vectors1,2 or plasmids3,4 for Cas9 and gRNA expression, which resulted in low targeting efficiency and high toxicity. These expression systems also pose concerns about safety in clinical translation due to the risk of unwanted genetic mutations and immunogenicity.5 More recently, electroporation of activated T cells with ribonucleoprotein (RNP) complexes made from recombinant Cas9 protein and in vitro-transcribed (IVT) or synthetic gRNAs has achieved high efficacy across a number of targets.6-10 In many cell types, including human T cells, IVT gRNAs cause high cytotoxicity due to the presence of a 5’ triphosphate single-stranded RNA (ssRNA) that activates a type I interferon-mediated immune response.11 Synthetic gRNAs are therefore a preferred format for the RNP-based method. ArciTect™ is an RNP-based CRISPR-Cas9 expression system that includes custom synthetic gRNA (sgRNA and crRNA) and is designed to fully support genome editing of human primary T cells.
Beyond CRISPR-Cas9 expression methods, the culture systems for expansion and activation of primary human T cells also represent critical elements for successful genome editing, with cell activation being required in most experimental contexts.12 While T cells can be isolated from a number of sources using a variety of isolation techniques, to date most genome editing studies involving T cells have used primary human T cells isolated from peripheral blood mononuclear cells (PBMCs) by immunomagnetic selection.7, 12-14
With improved expression and delivery methods, CRISPR-Cas9 genome editing is now rapidly being incorporated into the development of next-generation immunotherapies. One example is chimeric antigen receptor (CAR) T cell therapy, wherein CRISPR-Cas9 enables generation of allogeneic immune effector cells that are compatible for patient infusion.15 For this purpose, T cells can be engineered to escape immune rejection through deletion of endogenous T cell receptors (TCRs) and human leukocyte antigen (HLA) class I molecules, which shows high efficacy when combined with deletion of inhibitory receptors such as programmed death-1 (PD-1) or cytotoxic T-lymphocyte antigen protein 4 (CTLA-1).9-10,16-17 This strategy can then be coupled with targeting CAR constructs to the endogenous TCR alpha constant (TRAC) locus18 to generate universal allogeneic CAR-T cells.18
CRISPR-Cas9 Genome Editing in Primary Human T Cells
Materials Required
or
ArciTect™ crRNA†
ArciTect™ tracrRNA Kit
- ArciTect™ tracrRNA
- ArciTect™ Annealing Buffer (5X)
ImmunoCult™ Human CD3/CD28/CD2 T Cell Activator
- Resuspension Buffer T
- Electrolytic Buffer E2
- 10 μL Neon® pipette tips
or
P3 Primary Cell 4D-Nucleofector™ X Kit S
- 16-well Nucleocuvette™ Strips
- P3 Primary Cell Nucleofector™ Solution
- Supplement 1
or
Lonza V4XP-3032
†ArciTect™ sgRNA is only available in Australia, Austria, Belgium, Canada, China, Denmark, Finland, France, Germany, Iceland, Ireland, Luxembourg, the Netherlands, New Zealand, Norway, Poland, Portugal, Singapore, Spain, Sweden, Switzerland, the United Kingdom, and the United States.
A. T Cell Isolation and Activation
- Isolate human T cells from peripheral blood (e.g. Catalog #70500) using EasySep™ Human T Cell Isolation Kit. Refer to the Product Information Sheet (Document #DX20004) for details.
Optional: Source frozen primary T cells (e.g. Catalog #70024). Refer to the Product Information Sheet (Document #DX27569) for details. - Count cells and adjust to 1 x 106 cells/mL in ImmunoCult™-XF T Cell Expansion Medium supplemented with 2mM L-Glutamine, 50µg/mL gentamicin, and 10 ng/mL (130 IU/mL for the specific lot*) Human Recombinant IL-2.
*International Units (IU) need to be calculated per lot. For more information visit www.stemcell.com/international-units-conversion-data-for-cytokines. - Activate human T cells by adding 25 µL/mL of ImmunoCult™ Human CD3/CD28 T Cell Activator. Incubate cell suspension at 37°C and 5% CO2 for 72 hours.
- Optional: Assess T cell activation by binding the activation marker CD25 with Anti-Human CD25 Antibody, Clone BC96 (Catalog #60158) and performing flow cytometry.
B. Preparation of ArciTect™ sgRNA working solution and ArciTectTM crRNA and ArciTectTM tracrRNA stock Solutions
- Briefly centrifuge the vials before opening.
- Add nuclease-free water to each vial to give a final concentration of 200 μM (crRNA and tracrRNA) or 100μM (sgRNA), as indicated in Table 1 and Table 2, respectively.
- Mix thoroughly. If not used immediately, aliquot and store at -20°C for up to 6 months or at -80°C for longer than 6 months. After thawing the aliquots, use immediately. Do not refreeze.
Table 1. Preparation of 200 µM* ArciTect™ crRNA or ArciTect™ tracrRNA
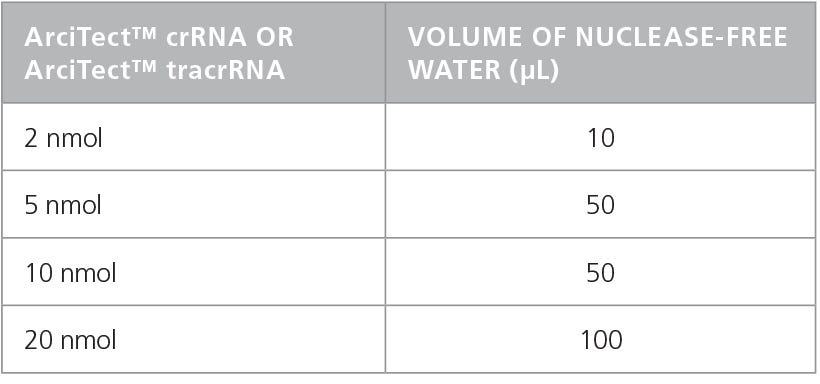
Table 2. Resuspension Volume for 100 µM* ArciTect™ sgRNA

C. Annealing of crRNA:tracrRNA Duplexes
RNP complexes can be prepared using either the two-part crRNA:tracrRNA gRNA, which requires pre-annealing (see below), or sgRNA. If working with sgRNA, proceed directly to section D.
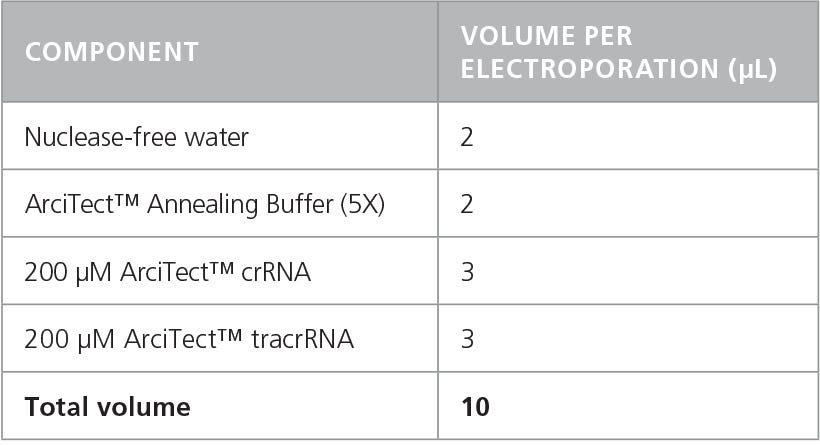
- Prepare a 60 μM crRNA:tracrRNA solution by combining components in a DNase- and RNase-free microcentrifuge tube as indicated in Table 3. Volumes are sufficient for a single electroporation reaction; adjust as required. Mix thoroughly.
- In a thermocycler or heating block, incubate crRNA:tracrRNA mixture at 95°C for 5 minutes followed by 60°C for 1 minute. Cool to room temperature (15 - 25°C) and place on ice. If not used immediately, store at -80ºC for up to 1 month.
Table 3. Preparation of 60 μM (60 pmol/μL) crRNA:tracrRNA Duplex
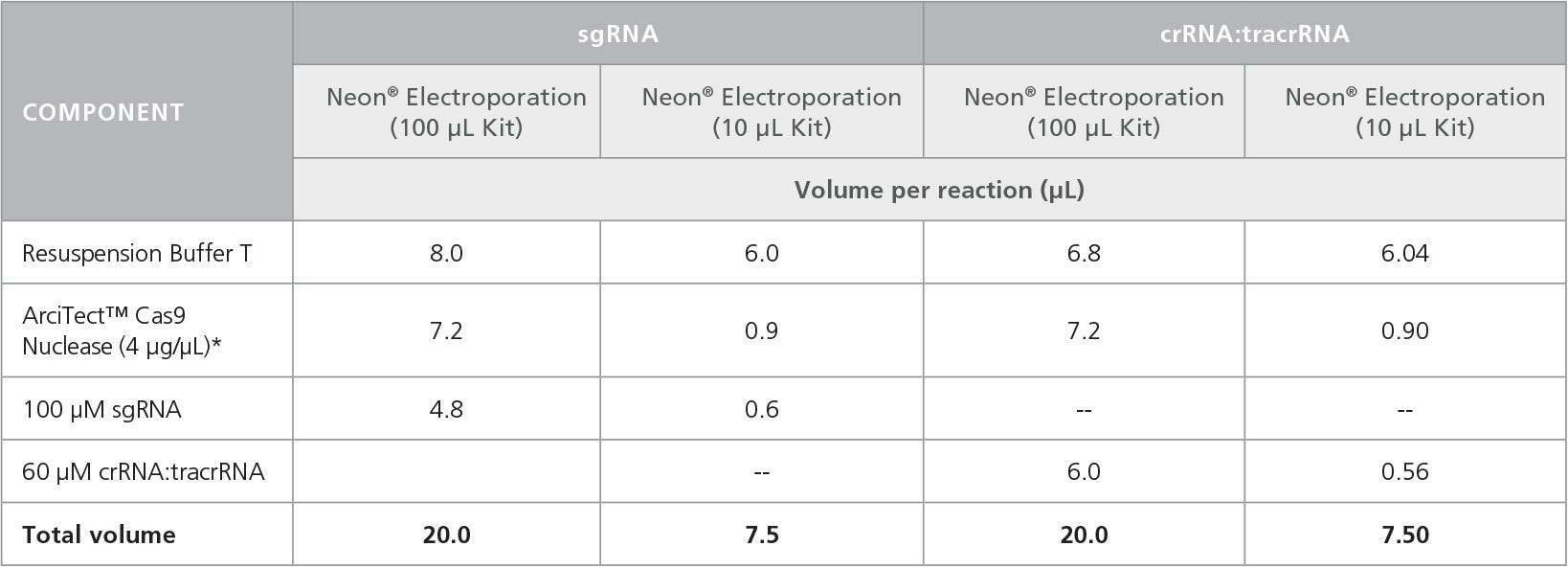
D. Preparation of ArciTect™ CRISPR-Cas9 RNP Complex Mix
- To prepare the RNP Complex Mix, combine the components in the order listed in Table 4 in a microcentrifuge tube. Adjust component volumes according to the desired number of transfections.
- Mix thoroughly.
- Mix the RNP Complex Mixture by gently pipetting up and down 2 times; avoid creating air bubbles or foam in the tube.
- Incubate the RNP Complex Mix at room temperature (15 - 25°C) for 10 - 15 minutes.
Table 4. Preparation of RNP Complex Mixture for Electroporation
E. Preparation of T Cell Suspension for Electroporation
- For each electroporation condition (including positive and negative controls), prepare 2 mL of ImmunoCult™-XF T Cell Expansion Medium supplemented with 2 mM L-Glutamine, 50 μg/mL gentamicin, and 10 ng/mL Human Recombinant IL-2.
- For each condition, add 2 mL of supplemented medium (prepared in step 1) to 1 well of a 6-well plate (for 100 μL tip reactions) or 1 mL of supplemented medium to 1 well of a 12-well plate (for 10 μL tip reactions) and place in a 37°C incubator. Store the remainder of the supplemented medium at 2 - 8°C.
- Transfer 1.2 x 106 cells (for 100 μL tip reactions) or 1.5 x 105 cells (for 10 μL tip reactions) from the activated T cell suspension (prepared in section A) to a 15 mL conical tube. Centrifuge at 300 x g for 5 minutes at room temperature.
F. Electroporation of T cells with RNP Complex Using Neon® Transfection System
- Aspirate supernatant from cell pellet (prepared in section E). Resuspend cells in 100 μL of Resuspension Buffer T, if using 100 μL tips, or 7.5 μL of Resuspension Buffer T, if using 10 μL tips, and pipette up and down to mix.
- Using individual RNase-free microcentrifuge tubes for each condition, combine RNP Complex Mixture (prepared in section D) with activated T cells as indicated in Table 5.
- Mix gently by pipetting up and down carefully 2 times; avoid creating air bubbles or foam in the tube.
Note: If air bubbles are present in the tip when the cells are electroporated, cell viability and transfection efficiency will be significantly reduced. - Use a 100 μL Neon® pipette tip to draw up 100 μL of the mixture and place into the electroporation chamber containing 3 mL of Electrolytic Buffer E2.
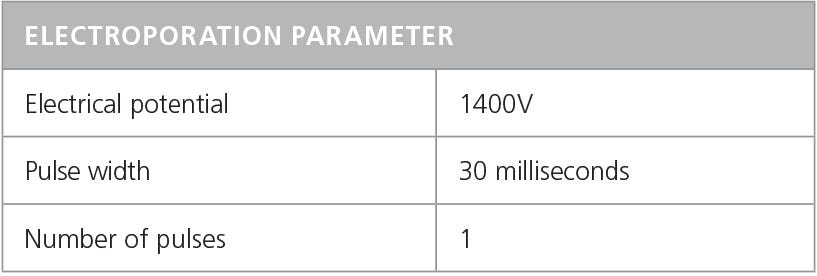
- Electroporate mixture according to conditions indicated in Table 6.
Note: Refer to the manufacturer’s instructions for electroporation. Electroporation conditions may require optimization for different cell types.
Table 5. Mixture of RNP Complex and T Cells for Electroporation
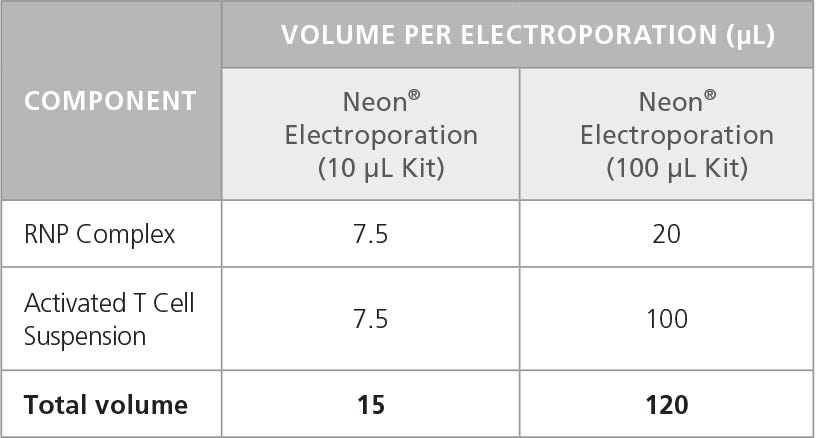
Table 6. Recommended Electroporation Conditions for Human T Cells Using a Neon® Transfection System
G. Post-Editing Culture
- Immediately add the electroporated cells to 1 well of the 6- or 12-well plate prepared in Section E step 2. Incubate at 37°C and 5% CO2 for 2 - 3 hours.
- Count cells and adjust cell density to 2.5 x 105 viable cells/mL by adding IL-2-supplemented ImmunoCult™-XF T Cell Expansion Medium (prepared in step 1).
- Incubate at 37°C and 5% CO2 for 48 - 72 hours for genome editing to occur. Harvest cells for assessment of genome editing efficiency, genomic DNA can be amplified by PCR using primers flanking the target region and ArciTect™ High-Fidelity DNA Polymerase Kit (Catalog #76026), followed by sequencing of PCR products. Alternatively, ArciTect™ T7 Endonuclease Kit (Catalog #76021) can be used to assess editing efficiency (% INDEL formation) following PCR amplification. For further information, refer to the Technical Bulletin: Evaluation of Genome Editing.
Case Study: Evaluation of Optimal Culture Methods for High Efficiency TRAC Knockout in Human Primary T Cells
To demonstrate genetic knockout with the ArciTect™ CRISPR-Cas9 system, we targeted the TRAC locus using the protocol described in this document. In addition, we tested multiple T cell activation reagents and dynamics to identify a condition with the highest knockout efficiency. Loss of function due to INDELs at this locus is readily identifiable by the lack of TCR expression on the cell surface. The practical importance of targeting this locus lies in the generation of universal allogeneic T cells for CAR T cell therapies.18
We first designed multiple crRNAs (Figure 3A, D), as different crRNA sequences can exhibit varying efficiency at the target site (see Technical Bulletin: Genome Editing with Direct Cas9 RNP Delivery Design Considerations). We tested the efficacy of the four crRNAs in activated human T cells, with TRAC knockout efficiency detected by flow cytometry, and INDEL generation detected by the ArciTect™ T7 Endonuclease I Kit. All four crRNAs significantly reduced the frequency of TCRαβ+ cells (Figure 3B) and resulted in cut bands in the T7 endonuclease I assay (Figure 3C), confirming the presence of genomic mismatches that are indicative of INDELs. Based on these results, we proceeded with crRNA2, as it showed the highest knockdown efficiency by flow cytometry.
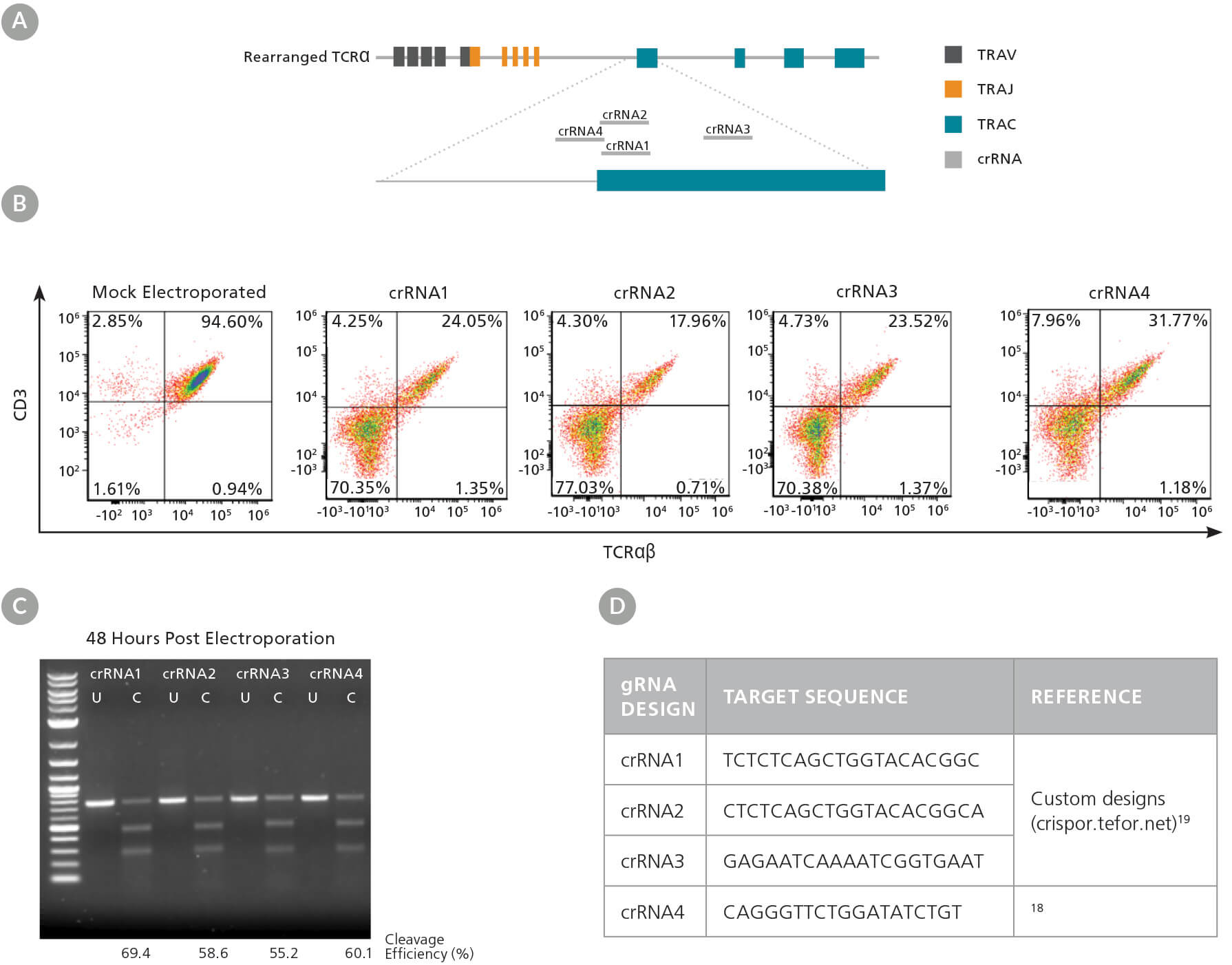
Figure 3. TRAC gRNA Design and Evaluation
(A) Schematic of the TRAC locus with crRNA sequences 1 - 4 aligned to the TRAC first exon. (B) Representative TCRαβ/CD3 flow cytometry results at 48 hours post RNP complex electroporation (containing the corresponding crRNA1 - 4) into activated T cells. (C) Genome editing (cleavage) efficiency was assessed at 48 hours post electroporation using the ArciTect™ T7 Endonuclease I Kit. U: Uncut; C: Cut: (D) gRNA target sequences for TRAC knockout.
We then used the validated crRNA2 to compare knockout efficiency in a variety of T cell activation conditions. First, T cells were isolated from the human peripheral blood of four independent donors. Cells were cultured in Immunocult™-XF T Cell Expansion Medium supplemented as described with either ImmunoCult™ Human CD3/CD28 T Cell Activator or CD3/CD28/ CD2 T Cell Activator for 2 or 3 days, for a total of 4 test conditions. On day 0, approximately 10 - 20% of cells expressed the activation marker CD25 (data not shown), which increased to over 80% after 2 or 3 days in the presence of the activators (Figure 4). T cell activation was greater when cells were incubated with the CD3/CD28/CD2 T Cell Activator compared to the CD3/CD28 T Cell Activator (Figure 4). The percentage of activated cells was also greater when cells were incubated with either activator for 3 days compared to 2 days (Figure 4). For more information about optimization of T cell activation and expansion conditions, refer to the Technical Bulletin: Optimization of Human T Cell Expansion Protocol: Effects of Early Cell Dilution.
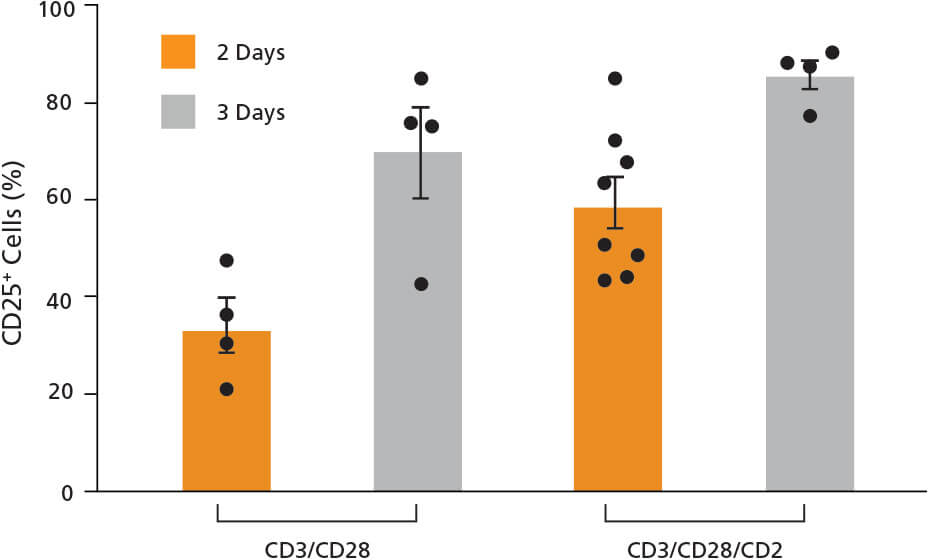
Figure 4. T Cells Express the CD25 Activation Marker After Treatment with ImmunoCult™ Human T Cell Activators
Isolated human T cells were activated with either ImmunoCult™ Human CD3/CD28 or CD3/CD28/CD2 T Cell Activator for 2 or 3 days. Activation status was assessed by CD25 flow cytometry. Each data point per condition represents an individual donor; n = 4 - 8 donors. Error bars represent standard error of the mean.
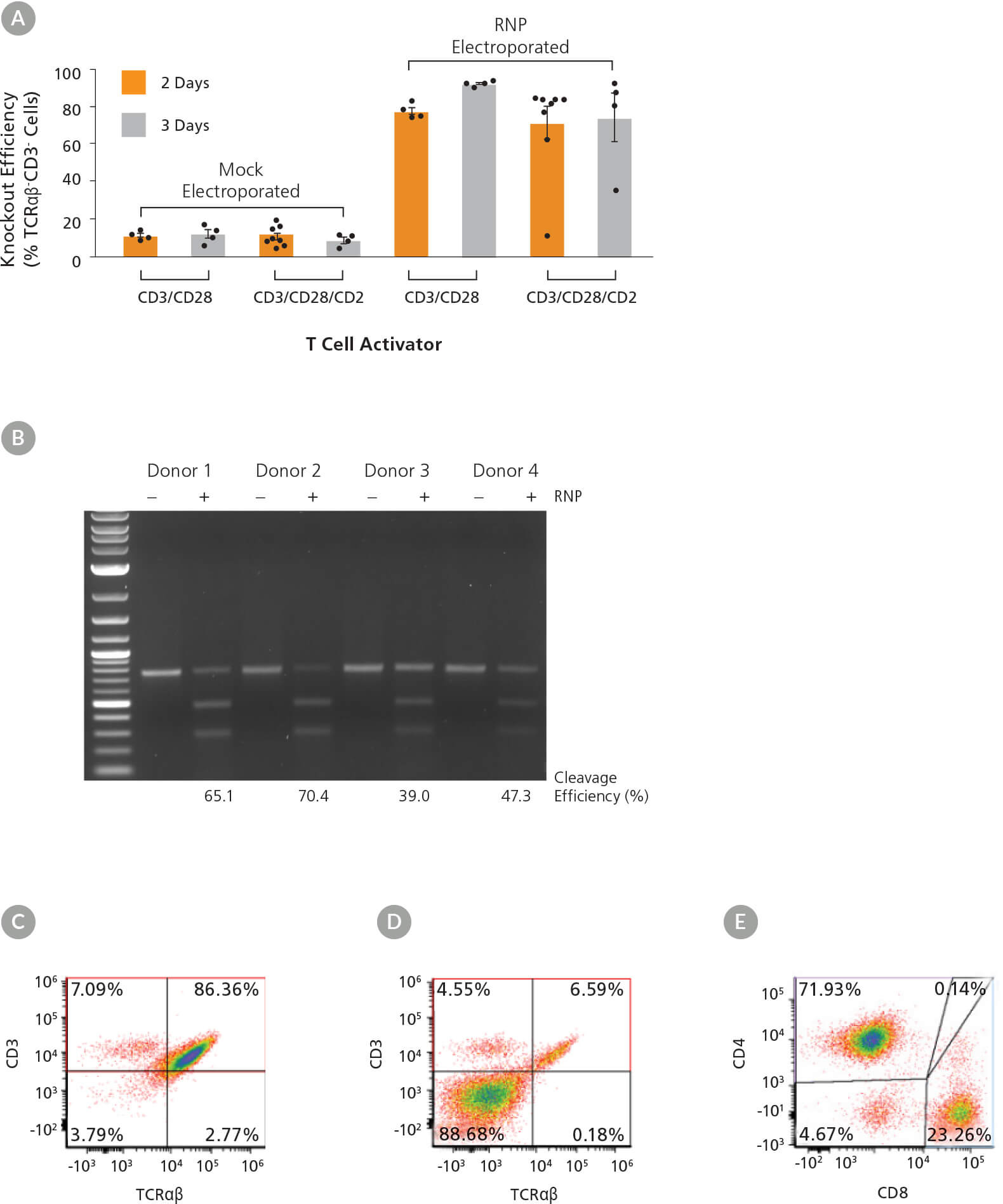
Figure 5. High Efficiency TRAC Knockout Across Activation Conditions and Dynamics
(A) TRAC knockout efficiency in human T cells activated with either ImmunoCult™ Human CD3/CD28 or CD3/CD28/CD2 T Cell Activator for 2 or 3 days was assessed by binding the TCRαβ and CD3 receptors with antibodies followed by flow cytometry analysis. Each data point per condition represents an individual donor; n = 4 - 8 donors. Error bars represent standard error of the mean. (B) Genome editing (cleavage) efficiency was assessed at 48 hours post electroporation in human T cells activated with ImmunoCult™ Human CD3/CD28 T Cell Activator for 3 days using the ArciTect™ T7 Endonuclease I Kit. Mock electroporated: - RNP; RNP electroporated: + RNP. (C - D) Representative dot plots of TCRαβ and CD3 flow cytometry analysis from (C) mock electroporated and (D) RNP electroporated human T cells activated with ImmunoCult™ Human CD3/CD28 T Cell Activator for 3 days. (E) Representative dot plot of CD4 and CD8 flow cytometry analysis of human T cells activated with ImmunoCult™ Human CD3/CD28 T Cell Activator for 3 days.
Following activation, T cells were electroporated with RNP complexes containing TRAC crRNA2. Viability ranged from 52 - 82% (data not shown) immediately after electroporation, independent of activation condition. Knockout efficiency was measured 3 days after electroporation (day 5 or 6 from start of activation), and we observed up to 90% increase in knockout efficiency (TCRαβ- cells; Figure 5A). The highest consistent knockout efficiencies were observed when cells were electroporated after 3 days of activation, particularly when the ImmunoCult™ Human CD3/CD28 T Cell Activator was used (Figure 5A). Moreover, INDEL formation at the target locus was confirmed by the T7 endonuclease I assay in treated samples from all 4 donors (Figure 5B). Results were more variable among donors when cultured with the ImmunoCult™ Human CD3/CD28/CD2 T Cell Activator over 2 days, with one non-responsive sample. We therefore tested an additional 4 donors with that condition and observed more consistent knockout in all additional samples. As TCRαβ forms a complex with CD3, the knockout was confirmed by concomitant loss of CD3 expression on the cell surface (Figure 5A, C, D), while T cell markers CD4 and CD8 continued to be highly expressed (Figure 5E).
Human primary T cells were then electroporated with RNP complexes containing sgRNA or crRNA:tracrRNA duplexes targeting TRAC (Figure 6A) or a second target, β2 microglobulin (B2M; Figure 6B), to test knockout efficiency with different gRNA formats. Since expression of B2M is required for surface expression of major histocompatibility class I (MHC-I) molecules, this strategy enabled quantitative readout of editing efficiency by flow cytometry to detect MHC-I expression.20,21 Both gRNA formats resulted in similar knockout efficiencies at either target, suggesting that the ArciTect™ CRISPR-Cas9 system exhibits high editing efficiency in human primary T cells regardless of gRNA formats. Both formats enable high editing efficiency.
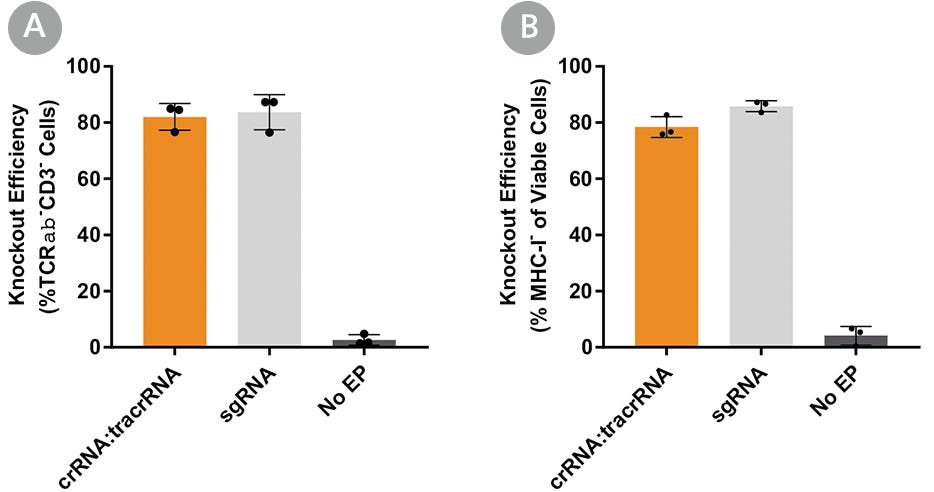
Figure 6. Comparison of TRAC and B2M Knockout Editing Efficiency Using Either ArciTect™ sgRNA or crRNA:tracrRNA Formats
Human T cells were activated with ImmunoCult™ Human CD3/CD28 T Cell Activator for 3 days and electroporated with CRISPR-Cas9 RNP complexes containing ArciTect™ Cas9 Nuclease and either ArciTect™ crRNA:tracrRNA duplexes or sgRNA targeting (A) TRAC or (B) B2M (B). Knockout efficiency was measured by flow cytometry 3 days after electroporation. Each data point per condition represents an individual donor; n = 3 donors. No EP: non-electroporated cells. Error bars represent standard deviation.
Following CRISPR-Cas9 genome editing, cells were cultured for an additional 10 days by adding in fresh Immunocult™-XF T Cell Expansion Medium supplemented with IL-2 every 2 - 3 days. TCRαβ- cells persisted over the course of expansion (data not shown). Cells treated with ImmunoCult™ Human CD3/CD28/CD2 T Cell Activator prior to electroporation exhibited a higher degree of expansion post gene editing compared to those treated with ImmunoCult™ Human CD3/CD28 T Cell Activator. Based on the fold expansion, calculated cell yields ranged from 4.1 x 107 to 3.8 x 108 cells after 7 days of incubation post-electroporation.
TCRαß-knockout efficiency typically ranged from 60 - 90% (Figure 5A). There has been a general interest in targeting TRAC to deplete TCRαß-expressing cells for the generation of allogeneic immune effector cells18, the success of which will require highly pure populations of TCRαß- cells. Figure 7 shows an example where the EasySep™ Human TCR Alpha/Beta Depletion Kit (Catalog #17847) was used to reduce the frequency of CD3+TCRαß+ cells from 19.1% to 0.1%.
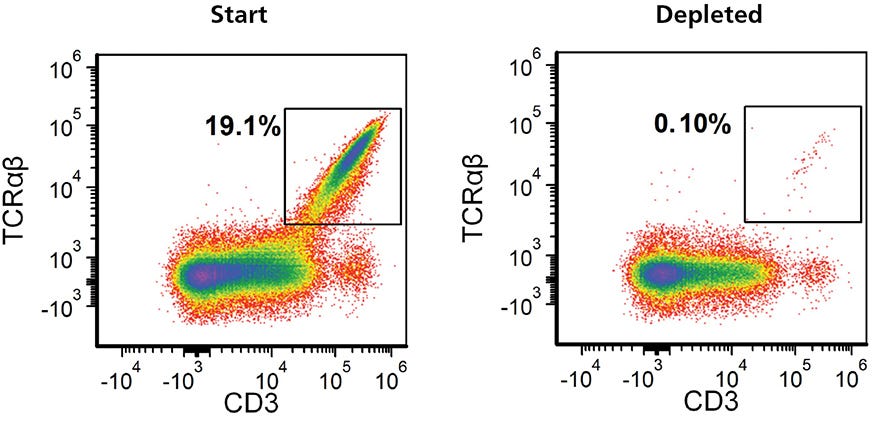
Figure 7. Residual TCRαβ+ Cells Can Be Removed from Expanded TRAC Knockout Cell Populations Using EasySep™ Human TCRαβ Depletion Kit
Gene-edited TCRαβ knockout T cells were expanded for 7 - 10 days, harvested and resuspended in EasySep™ Buffer at 5 x 107 cells/mL. Following a 13-minute EasySep™ TCRαβ depletion protocol, flow cytometry was performed to assess the depletion of TCRαβ+ cells. Representative data plots are shown for the frequency of TCRαβ cells in the expanded TRAC kncockoutcell population before and after EasySep™ depletion.
Discussion
In editing primary human T cells, there exists a delicate balance between cell exhaustion, cell viability, and gene editing efficiency. We found
that stimulation with ImmunoCult™ Human CD3/CD28 T Cell Activator for 3 days prior to electroporation resulted in the highest and most
consistent editing efficiencies. Greater cell expansion post editing was observed for cells stimulated with ImmunoCult™ Human CD3/CD28/CD2 T
Cell Activator; consider the output cell number required for downstream applications when choosing a protocol. If a greater number of edited
cells are required for downstream applications, one can also scale up with more cells per electroporation or with more wells. In general, two
wells are recommended for positive (e.g. ArciTect™ Human HPRT Positive Control Kit, Catalog #760134) and negative (non-electroporated)
controls. The total number of wells should be determined before starting, to calculate the total amount of materials required. Optimization of
conditions such as cell density or Cas9:gRNA ratio may be necessary, or multiple donor samples may be required (e.g. diseased and healthy).
The genome editing strategy outlined in this document is typical for most genetic knockout applications. Application-specific protocol
modifications, not thoroughly detailed here, might include: use of Cas9-eGFP Nuclease for visualization of positive transfectants; use of the single-strand endonuclease Cas9 Nickase with two flanking gRNAs; or the addition of a DNA donor template for homology-directed repair (HDR)-mediated genetic knock-in.

Production of CAR T Cells Wallchart
This free Nature Protocols Wallchart summarizes the processes involved in producing CAR T cells for therapy, including cell enrichment, activation and expansion.
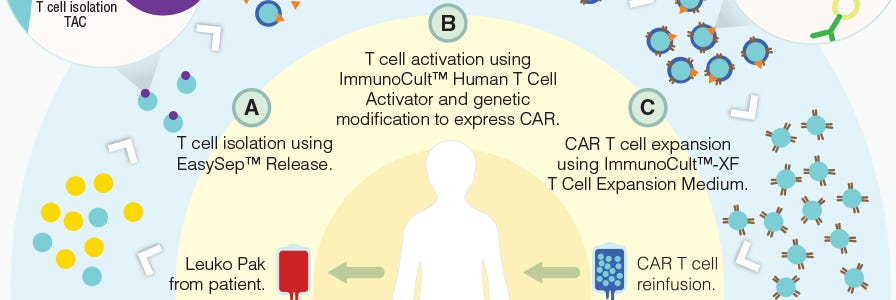
T Cell Therapy Reagents
Explore tools for your T cell therapy research, including products for T cell isolation, activation, and expansion.
Additional Resources



References
- Wang W et al. (2014) CCR gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS One 9:e115987.
- Li C et al. (2015) Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J Gen Virol 96:2381–93.
- Mandal PK et al. (2014) Efficient ablation of genes in hematopoietic stem and effector cells using CRISPR/Cas9. J Gen Virol 163: 1817–26.
- Su S et al. (2016) CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep 6: 20070.
- Yin H et al. (2014) Non-viral vectors for gene-based therapy. Nat Rev Genet 15: 541–55.
- Hendel A et al. (2015) Chemically modified guide RNAs enhance CRISPR-Cas9 genome editing in human primary cells. Nat Biotechnol 33: 985–9.
- Schumann K et al. (2015) Generation of knock-in primary human T cells using Cas9 ribonucleoprotein. Proc Natl Acad Sci USA 112: 10437–42.
- Gomes-Silva D et al. (2017) CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 130: 285–96.
- Ren J et al. (2017) Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res 23: 2255–66.
- Rupp LJ et al. (2017) CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep 7(1): 737.
- Kim S et al. (2018) CRISPR RNAs trigger innate immune responses in human cells. Genome Res 28: 1–7.
- Seki A & Rutz S (2018) Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med 215(3): 985.
- Hou P et al. (2015) Genome editing of CXCR4 by CRISPR/Cas9 confers cells resistant to HIV-1 infection. Sci Rep 5: 15577.
- Hultquist JF (2016) A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human Cells. Cell Rep 17: 1438–52.
- Jung I-Y & Lee J (2018) Unleashing the therapeutic potential of CAR-T cell therapy using gene-editing technologies. Mol Cells 41(8): 717–23.
- Liu X et al. (2017) CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res 27: 154–7.
- Ren J et al. (2017) A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 8: 17002–11
- Eyquem J et al. (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543(7643): 113–7.
- Haeussler M et al. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17: 148.
- Bjorkman PJ et al. (1987) Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329: 506–12.
- Zijlstra M et al. (1990) Beta 2-microglobulin deficient mice lack CD4-8+ cytolytic T cells. Nature 344: 742–6.

Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration

