CRISPR Guide Design Tools and Algorithm
The CRISPR Guide Design Tool uses best practices and the latest computational tools to deliver the optimal CRISPR RNA (crRNA) or single guide RNA (sgRNA) sequence for every gene in the human and mouse genomes.
Access the CRISPR Design Tools
CRISPR Guide Design Algorithm
A large number of candidate guide sequences exist for each gene (up to 1000’s for certain genes), but determining the optimal guide sequence to choose is a very difficult task. Many requirements must be considered when picking the guide sequence to use for knocking out a specific gene. The current algorithm considers the requirements described below, then generates scores for each one. Targets are then ranked and the top ones for each gene are selected.
Below, each step of our guide search and ranking algorithm is described with rationale and methodology provided where applicable.
Target Ranking
All candidate targets with the capability of knocking out a gene, i.e. the cut-site of the target overlaps the coding region of a particular gene, are extracted from the target genome. For instance, we identified 9,967,686 guide sequences that target coding regions in the human genome (GRCh38).
The targets for each gene are first sorted based on specific parameters passing certain thresholds, with those that pass being given the highest priority. Parameters include on- and off-target score, relative position within the gene, single-nucleotide polymorphism (SNP) probability, and fraction of isoforms covered (descriptions of each parameter and scoring methodology are given in the next section: Parameter Scoring) passing certain thresholds, with those that pass being given the highest priority. The threshold for each parameter is:
- On-target score ≥ 0.4
- On-target score ≥ 0.4
- Off-target score ≥ 0.67
- Relative target position ≤ 0.5
- SNP probability ≤ 0.05
- Fraction covered > 0.5
Note: In some cases, no guides for a particular gene meet all the thresholds. In these instances, the importance of passing thresholds is ranked as follows:
SNP probability > fraction covered > relative target position > off-target score > on-target score
Top targets for specific isoforms are also generated. These use all of the above criteria except fraction covered, as that is no longer relevant. To get the optimal targets for a specific isoform, the customer should enter the RefSeq isoform name, rather than the gene name. Note: Only verified mRNA and ncRNA (MN and NR) transcripts are included. Predicted mRNA and ncRNA (XM and XR) are not counted in the proportion of isoforms a particular guide will target.
A weighted average overall target score (range 0 - 1) is calculated using the scores assigned to on-target strength, off-target capability, relative position within the gene, and the fraction of transcripts covered. The weighting for each parameter is:
- Relative target position: 0.4
- Fraction covered: 0.4
- On-target score: 0.1
- Off-target score: 0.1
Parameter Scoring
On-target Strength
Targeting specificity of gRNAs is determined by complementarity between the guide sequence and a corresponding genomic DNA sequence. In order for a double-stranded break (DSB) to occur at the guide-specified location, a strong interaction between the guide sequence and the complementary DNA sequence must occur. Depending on this strength, the probability of successful DSB formation varies.
An on-target score is generated for every target (score between 0 - 1), with a higher score indicating a stronger on-target strength. The algorithm used to determine on-target scores can be found in Doench et al. (1).
Off-target Capability
Ideally, a particular guide will have 100% homology with the target sequence and no homology elsewhere in the genome. However, as target sequence binding can tolerate several mismatches, there often exist many potential off-target sites that contain one or more mismatches.
An off-target score is generated (between 0 - 1) that indicates the inverse probability of off-target cutting, with a higher score denoting targets with lower off-target potential.
Relative Target Position within Gene
Targets with a cut site closer to the N-terminus of a gene have a greater probability of resulting in functional genetic knockout. This is because a frameshift at the start of a gene will disrupt a greater proportion of the protein than a frameshift at the end.
The relative target position is scored relative to the beginning of the coding region for protein-coding transcripts (range 0 - 1), where a lower score indicates a guide closer to the N-terminus of the gene.
SNP Probability
Mismatches between the target and the guide can significantly hinder the interaction strength; even just a single mismatch can significantly reduce interaction strength between the guide sequence and complementary genomic sequence, and result in reduced cutting/editing efficiency.
SNP probabilities (range 0 - 1) indicate the likelihood of at least one base variation in the target sequence. The probability of the target harboring an SNP is based on the number of SNPs found within the target sequence, and the allele frequency of the SNP within the population.
Note: SNP probability is not included for mouse guides, as mice are typically inbred.
Fraction of Transcripts Covered
Many genes encode multiple isoforms. Unless a specific isoform is indicated as the gene target in the design tool, guides that target all or the majority of isoforms are preferred. Only verified mRNA and ncRNA (MN and NR) transcripts are included. Predicted mRNA and ncRNA (XM and XR) are not counted in the proportion of isoforms a particular guide will target.
CRISPR Genome Editing Products and Resources

ArciTect™ CRISPR-Cas9 Genome Editing System
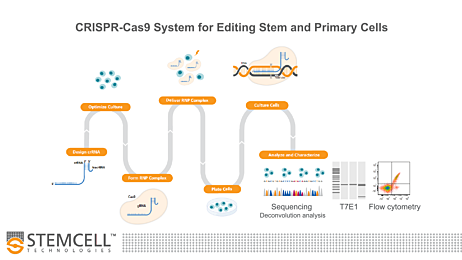
Overcome challenges in efficient delivery and expression of CRISPR machinery using an RNP-based CRISPR-Cas9 system. ArciTect™ enables researchers to perform high-efficiency genome editing in difficult-to-manipulate cell types, including stem and primary cells.

Wiley E-Book: Genome Editing Applications
Learn about next-generation disease modeling using CRISPR, including comprehensive genome editing strategies for complex cell culture models, suggestions for optimizing experimental conditions, and troubleshooting tips.



Resources
- Crisflash to extract all guide locations and identify off-targets (PMID: 30649181) (2).
- Azimuth to determine ontarget scores (PMID: 26780180)(1).
- Genome Assembly & RefSeq annotations based on: GRCh38.p13 / GRCm38.p6.
- SNPs identified using locations and allele frequencies from dbSNP build GCF_000001405.38.
- All other software was developed in house and is proprietary.
References
- Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34: 184–91.
- Jacquin ALS et al. Crisflash: open-source software to generate CRISPR guide RNAs against genomes annotated with individual variation. Bioinformatics 35(17): 3146–7.
