Genome Editing with Direct Cas9 RNP Delivery Design Considerations
- Document # 27083
- Version 1.0.1
- Jun 2018
Introduction
Originally discovered as a bacterial adaptive defense system, CRISPR-Cas9 uses antisense RNA remnants from past viral invasions combined with RNA-guided DNA cleavage to combat viral attack.1 The power of this system for targeted genome editing was quickly recognized. It has been widely adopted in many fields of cell biology research to generate targeted loss-offunction, gain-of-function, alternately regulated or protein-tagged gene mutations, whose function can then be studied in vitro or in model organisms.2 It also has therapeutic potential
to correct disease-causing mutations, particularly when coupled with regenerative medicine
using pluripotent stem cells. This system provides precisely targeted genome editing with high
efficiency and relatively low cost, and has been heralded as a major technological advance
toward clinical gene therapy.
The CRISPR-Cas9 system consists of RNA and protein components, which together form a ribonucleoprotein (RNP) complex (Figure 1). The
wildtype Cas9 protein has DNA endonuclease activity, and makes double-strand cuts upstream to a protospacer adjacent motif (PAM) sequence
in the target genome. S. pyogenes Cas9 recognizes the 3-nucleotide PAM site, NGG (where N is any nucleotide, followed by two guanines [G]),
and cleaves between the third and fourth nucleotides 5’ to the PAM site.
Table of Abbreviations
- Ribonucleoprotein
- CRISPR RNA
- Trans-activating crRNA
- Insertion or Deletion
- Protospacer Adjacent Motif
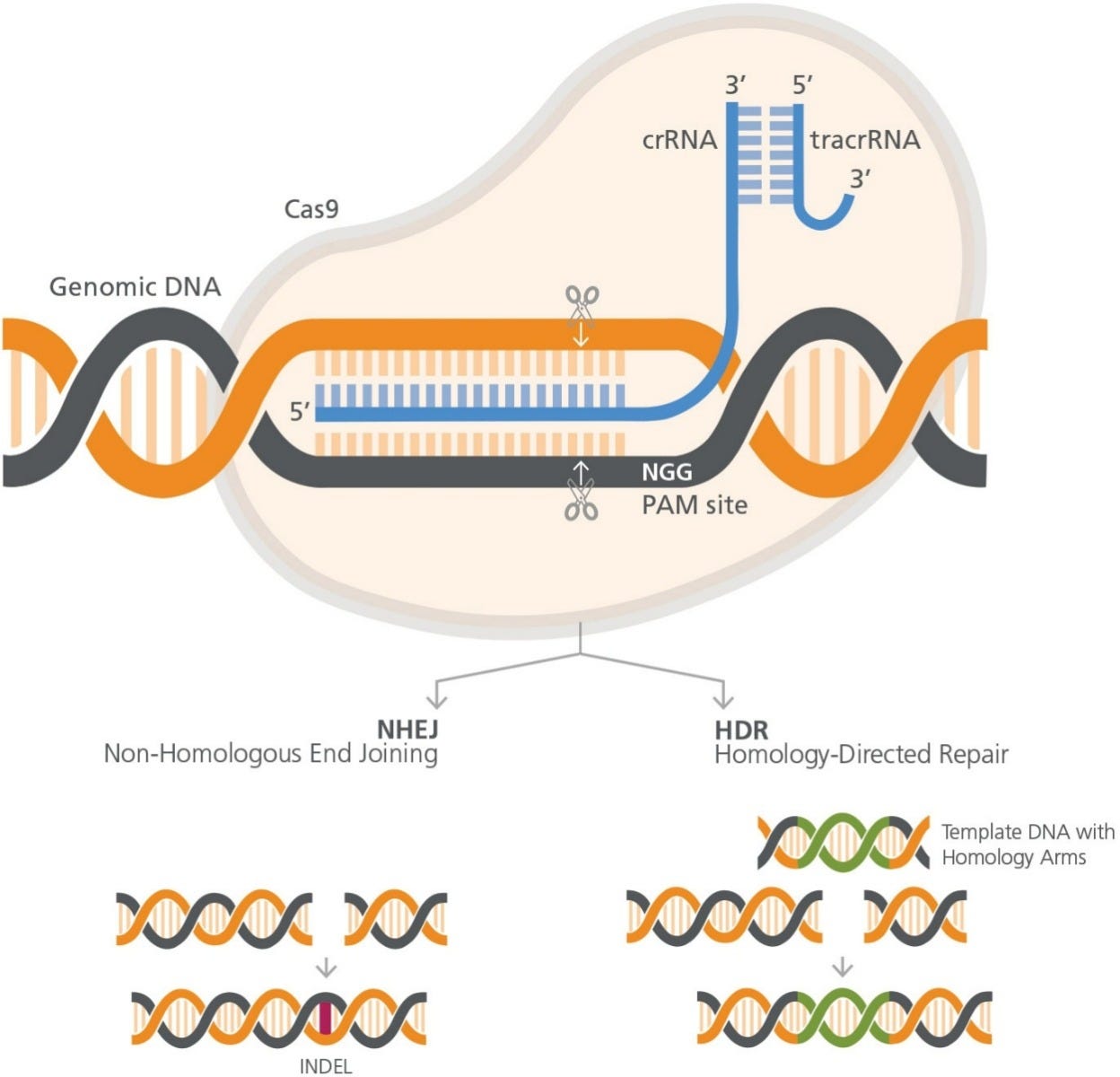
Figure 1. RNP Complex Orientation with respect to target cut site and PAM site
The RNA component is required for specific target recognition,
binding to an approximately 20 base pair complementary sequence
in the genomic DNA. Full guide RNA activity comes from a duplex
of two RNA molecules: a CRISPR RNA (crRNA) molecule, which
is complementary to the target, and a transactivating CRISPR
RNA (tracrRNA) molecule. The crRNA and tracrRNA molecules
form a duplex via a short homologous region. Association of the
crRNA:trRNA duplex with Cas9 causes a conformational change
in the nuclease, allowing it to bind DNA at the nearby PAM site
and implement the double-strand cut. Thus both complementarity
to the crRNA and an adjacent PAM site are required for targeting
Cas9-mediated cleaveage.
CRISPR-Cas9 genome editing has been widely adopted by the
research community, and application of the technology has
progressed rapidly. Previous CRISPR-Cas9 systems involved
transfection or transduction of multiple engineered plasmid or viral
vectors encoding the crRNA:tracrRNA and Cas9 components. The
vectors often integrated permanently into the genome, whereupon
their expression allowed the RNP to assemble inside the cell. These
systems were prone to insertional mutagenesis, prolonged CRISPRCas9
activity, and off-target mutational events.
Advances in recombinant protein and synthetic RNA production
have made it possible to directly utilize the crRNA and tracrRNA
molecules and the Cas9 protein. These can be formed into an RNP
in vitro and then transfected directly into the cell. This RNP system
is faster and easier to use, and results in less off-target events
compared to plasmid or viral systems due to the transient nature of
the editing molecules.
Types of Genome Editing
A variety of editing approaches can be employed to achieve
different types of mutational outcomes. Loss-of-function or
“knockout” mutations require complete destruction of gene
function, which can be obtained from a variety of possible
insertional or deletional events at the start of the coding sequence.
Conversely, corrective point mutations require extremely precise
changes. Knock-in mutations, often employed to mark genomic loci
and/or track expression patterns, require the addition of exogenous
DNA. In this section we summarize the different editing techniques
used and how their results vary.
Knockout mutations are typically generated by a process known as
non-homologous end joining (NHEJ). In this method, CRISPR-Cas9
is used to create a targeted a double-strand DNA break in the gene
of interest. The cell will repair the blunt-end break, using intrinsic
DNA repair processes. The repair processes are error-prone, and
often lead to the formation of small insertions or deletions (INDELs)
that can result in truncated proteins. Alternatively, one might use
CRISPR-Cas9 to generate double-strand breaks at two sites within
the gene, thereby creating a large deletion spanning the two targets. Targeting the first or second coding exon will help to ensure
complete loss of function.
NHEJ specificity can be increased through the use of Nickase
Cas9. In this form of the protein, one active domain has been
deactivated (via D10A mutation), resulting in only single-strand
endonuclease activity.3 Nickase Cas9 cuts only the DNA strand that
is complementary to the guide RNA. In order to affect a doublestrand
cut, a second guide RNA must be designed to target the
opposite strand. When two crRNAs in close proximity but targeting
opposite strands are used, the result is an overhanging doublestrand
cut. Because the probability of off-target sites with homology
to both of the two target sequences is vastly reduced, this method
produces fewer off-target mutational events. When Nickase is used,
the target cut sites should be 50-70 base pairs apart, on opposite
strands. The two crRNAs should be oriented so that their respective
PAM sites are facing towards the outside of the target region. This
will ensure double-strand breakage without RNP interference. While
the Nickase method offers advantages, it is limited in the availability
of two optimally spaced PAM sites at the target locus.
Homology directed repair (HDR) results in more finely-tuned
alterations. HDR employs exogenous DNA as a template to repair
the genome after CRISPR-Cas9-induced cleavage. The introduced
DNA includes stretches of homologous sequences, called homology
arms, flanking an exogenous sequence containing the desired
correction or genetic addition proximal to the targeted doublestrand
break. The exogenous sequence will be integrated at the
cleavage site, as the introduced DNA is used as a template during
the repair process.
Donor templates for HDR are selected based on the size of the
desired insertion: for large inserts such as fluorescent proteins or
selection cassettes, plasmids are typically used; for mutations of less
than 50 base pairs, synthetic single-stranded DNA oligonucleotide
(ssODN) templates can be utilized. With plasmids providing a
large double-stranded DNA donor template, each homology arm
should be at least 200 base pairs. Homology arms for short ssODN
templates should be no less than 40 base pairs, but ideally in the
range of 50 - 80 base pairs. Design the ssODN template with
homology arms so it is about 100 - 200 base pairs in length with
the Cas9 cut site at the center of the template.4,5
crRNA Design
The crRNA is a critical component of CRISPR-Cas9 genome editing
and requires careful design considerations as it will determine the
precise cut site of the Cas9 endonuclease.
crRNA sequences typically have 20 base pairs of sequence
homology with the intended genomic target, located immediately
adjacent to a PAM site. Since the frequency of NGG PAM sites
is approximately 5.21% in the human genome6, most genes will
have multiple potential crRNA target sequences. Typically, at least three independent crRNAs are tested in parallel to derive distinct edited populations in order to independently verify functional effects of the
mutation. This also increases the probability that at least one of the tested crRNAs effectively target Cas9 to the area of interest. Note that the
PAM sequence is not included within the crRNA.
As mentioned above, the intergenic location of the target site is an important consideration. If the gene contains multiple splice variants, a
constitutive exon should be targeted. If the goal is to generate a loss-of-function mutant, targeting the first coding exon is a common strategy.
Alternatively, creating larger deletions (>50 base pairs) by utilizing two crRNAs is an efficient way of disrupting gene function. The later method
also allows for easier identification of positive transfectants through PCR genotyping across the deletion. If modulation of gene expression is the
goal, target promoter elements within 200 base pairs of the transcription start site.
A number of online design tools perform comprehensive analysis of any target sequence, providing potential crRNA sequences and sites with
potential off-target activity (See Table 1). These tools identify PAM sites within the target sequence and compile a list of recommended crRNA
sequences that immediately precede the PAM sites. A genetic map of the crRNA’s intragenic location is typically included. crRNA sequences are
scored and ranked based on the number of, and the degree of homology to, potential off-target sites. Select crRNAs from the output list based
on location, homology score, and orientation, based on the guidelines described above.
Table 1. Online crRNA Design Tools
CRISPR-Cas9 is versatile and user-friendly compared to previous methods for precise genome editing. The ArciTect™ genome editing system represents a customizable, inexpensive tool for discovery and therapeutic application across all fields of life science research.
Further Resources
Addgene is a global, nonprofit repository that was created to help scientists share DNA-based research reagents, and offers a variety of
educational resources, including protocols, blog posts, and eBooks. The CRISPR 101 blog series were designed to help scientists of all levels
learn more about genome engineering: http://info.addgene.org/crispr-topic-page
This resource includes specific tips on how to design your crRNA for CRISPR genome editing: http://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing
- Jinek M et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816–21.
- Doudna JA & Charpentier E. (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213): 1258096.
- Ran FA et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6): 1380–9.
- Chen F et al. (2011) High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods 8(9): 753–5.
- Davis L & Maizels N. (2014) Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc Natl Acad Sci U S A 111(10): E924-32.
- Sherer S. (2008) A Short Guide to the Human Genome (pp. xiv, 173). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Haeussler M et al. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17(1): 148.
- Cradick TJ et al. (2014) COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol Ther Nucleic Acids 3: e214.
- Heigwer F et al. (2014) E-CRISP: fast CRISPR target site identification. Nat Methods 11(2): 122–3.
- Hsu PD et al. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9): 827–32.
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration

