Make more informed purchasing decisions with our new product availability and delivery estimate feature, now available on all product pages, in your cart, and during checkout.
Sign In
New to STEMCELL?
Register for an account to quickly and easily purchase products online and for one-click access to all educational content.
Thank you for your interest in this product.
Please provide us with your contact information and your local representative
will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
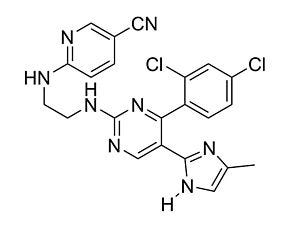
CHIR98014 is a reversible, cell-permeable activator of the WNT pathway, through inhibition of both isoforms of glycogen synthase kinase 3 (GSK3α and GSK3β) with IC₅₀ values of 0.65 and 0.58 nM, respectively. It shows at least 500-fold selectivity for GSK-3 versus 20 other serine/threonine or tyrosine kinases (Ring et al.).
DIFFERENTIATION
· Induces differentiation of endothelial progenitor cells from human induced pluripotent stem cells in the absence of VEGF (Lian et al.).
· Enhances osteogenic-like differentiation of rat mesenchymal stem cells cultured with high phosphate, including BMP-2 expression, calcium deposition, and alkaline phosphatase activity (Guerrero et al.).
METABOLISM
· Activates glycogen synthase in cells, lowers blood glucose levels and improves glucose disposal in insulin-resistant rats and diabetic db/db mice (Ring et al.).
DISEASE MODELING
· Reduces tau phosphorylation in the cortex and hippocampus of postnatal rat brains, which is implicated in the formation of neurofibrillary tangles and defects in axonal transport in Alzheimer’s disease (Selenica et al.).
This product is designed for use in the following research area(s) as part
of the highlighted workflow stage(s). Explore these workflows to learn more about the other products we
offer to support each research area.
Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small-molecule activation of WNT signaling.
Lian X et al.
Stem cell reports 2014 NOV
Abstract
Human pluripotent stem cell (hPSC)-derived endothelial cells and their progenitors may provide the means for vascularization of tissue-engineered constructs and can serve as models to study vascular development and disease. Here, we report a method to efficiently produce endothelial cells from hPSCs via GSK3 inhibition and culture in defined media to direct hPSC differentiation to CD34(+)CD31(+) endothelial progenitors. Exogenous vascular endothelial growth factor (VEGF) treatment was dispensable, and endothelial progenitor differentiation was β-catenin dependent. Furthermore, by clonal analysis, we showed that CD34(+)CD31(+)CD117(+)TIE-2(+) endothelial progenitors were multipotent, capable of differentiating into calponin-expressing smooth muscle cells and CD31(+)CD144(+)vWF(+)I-CAM1(+) endothelial cells. These endothelial cells were capable of 20 population doublings, formed tube-like structures, imported acetylated low-density lipoprotein, and maintained a dynamic barrier function. This study provides a rapid and efficient method for production of hPSC-derived endothelial progenitors and endothelial cells and identifies WNT/β-catenin signaling as a primary regulator for generating vascular cells from hPSCs.
TGF-β prevents phosphate-induced osteogenesis through inhibition of BMP and Wnt/β-catenin pathways.
Guerrero F et al.
PloS one 2014
Abstract
BACKGROUND: Transforming growth factor-β (TGF-β) is a key cytokine during differentiation of mesenchymal stem cells (MSC) into vascular smooth muscle cells (VSMC). High phosphate induces a phenotypic transformation of vascular smooth muscle cells (VSMC) into osteogenic-like cells. This study was aimed to evaluate signaling pathways involved during VSMC differentiation of MSC in presence or not of high phosphate. RESULTS: Our results showed that TGF-β induced nuclear translocation of Smad3 as well as the expression of vascular smooth muscle markers, such as smooth muscle alpha actin, SM22α, myocardin, and smooth muscle-myosin heavy chain. The addition of high phosphate to MSC promoted nuclear translocation of Smad1/5/8 and the activation of canonical Wnt/β-catenin in addition to an increase in BMP-2 expression, calcium deposition and alkaline phosphatase activity. The administration of TGF-β to MSC treated with high phosphate abolished all these effects by inhibiting canonical Wnt, BMP and TGF-β pathways. A similar outcome was observed in high phosphate-treated cells after the inhibition of canonical Wnt signaling with Dkk-1. Conversely, addition of both Wnt/β-catenin activators CHIR98014 and lithium chloride enhanced the effect of high phosphate on BMP-2, calcium deposition and alkaline phosphatase activity. CONCLUSIONS: Full VSMC differentiation induced by TGF-β may not be achieved when extracellular phosphate levels are high. Moreover, TGF-β prevents high phosphate-induced osteogenesis by decreasing the nuclear translocation of Smad 1/5/8 and avoiding the activation of Wnt/β-catenin pathway.
Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation.
Selenica M-L et al.
British journal of pharmacology 2007
Abstract
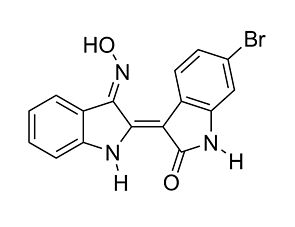
BACKGROUND AND PURPOSE: Glycogen synthase kinase-3 (GSK-3) affects neuropathological events associated with Alzheimeŕs disease (AD) such as hyperphosphorylation of the protein, tau. GSK-3beta expression, enzyme activity and tau phosphorylated at AD-relevant epitopes are elevated in juvenile rodent brains. Here, we assess five GSK-3beta inhibitors and lithium in lowering phosphorylated tau (p-tau) and GSK-3beta enzyme activity levels in 12-day old postnatal rats. EXPERIMENTAL APPROACH: Brain levels of inhibitors following treatment in vivo were optimized based on pharmacokinetic data. At optimal doses, p-tau (Ser(396)) levels in brain tissue was measured by immunoblotting and correlated with GSK-3beta enzyme activities in the same tissues. Effects of GSK inhibitors on p-tau, GSK-3beta activities and cell death were measured in a human neuronal cell line (LUHMES). KEY RESULTS: Lithium and CHIR98014 reduced tau phosphorylation (Ser(396)) in the cortex and hippocampus of postnatal rats, while Alsterpaullone and SB216763 were effective only in hippocampus. AR-A014418 and Indirubin-3'-monoxime were ineffective in either brain region. Inhibition of p-tau in brain required several-fold higher levels of GSK inhibitors than the IC(50) values obtained in recombinant or cell-based GSK-3beta enzyme activity assays. The inhibitory effect on GSK-3beta activity ex vivo correlated with protection against cell death and decrease of p-tau- in LUHMES cells, using low microM inhibitor concentrations. CONCLUSIONS AND IMPLICATIONS: Selective small-molecule inhibitors of GSK-3 reduce tau phosphorylation in vivo. These findings corroborate earlier suggestions that GSK-3beta may be an attractive target for disease-modification in AD and related conditions where tau phosphorylation is believed to contribute to disease pathogenesis.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.