Make more informed purchasing decisions with our new product availability and delivery estimate feature, now available on all product pages, in your cart, and during checkout.
Sign In
New to STEMCELL?
Register for an account to quickly and easily purchase products online and for one-click access to all educational content.
Thank you for your interest in this product.
Please provide us with your contact information and your local representative
will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
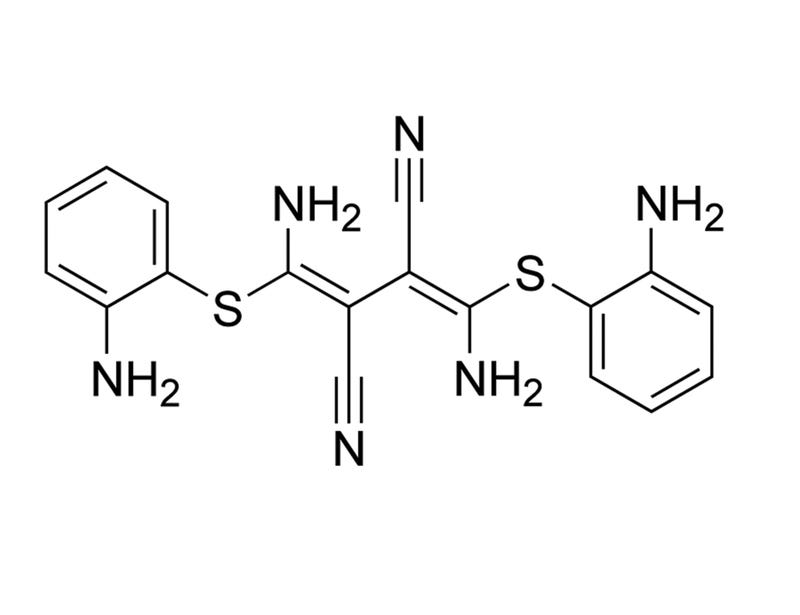
U-0126 is a selective, non-ATP competitive inhibitor of mitogen activated protein kinase kinase (MEK), inhibiting MEK1 and MEK2 with IC₅₀ values of 72 nM and 58 nM, respectively (Favata et al.; Scherle et al.). It shows little or no inhibition at micromolar levels of other kinases such as ERK, protein kinase C (PKC), c-Jun N-terminal kinases (JNK), other MAP kinase kinases (MKK3, MKK4, MKK6), cyclin-dependent kinases (CDK2, CDK4), ABL, and RAF (Favata et al. 1998). U-0126 also antagonizes AP-1 transcription and selectively inhibits promoters containing AP-1 response elements (Favata et al.).
MAINTENANCE & SELF-RENEWAL
· In combination with basic FGF, Activin A, and a PKC inhibitor, U-0126 promotes maintenance of human pluripotent stem cells (Kinehara et al.)
· Used alone with MEF-conditioned medium, U-0126 inhibits self-renewal of human pluripotent stem cells, leading to differentiation, without affecting proliferation or survival (Li et al.).
· Inhibits glutamate-induced oxidative stress in mouse hippocampal HT22 cells and rat primary cortical cultures (Satoh et al.; Ong et al.).
· Neuroprotective in a gerbil ischemia model and in primary mouse neurons cultured under hypoxia (Namura et al.).
IMMUNOLOGY
· Inhibits T-cell proliferation against certain antigenic stimuli by reducing IL-2 mRNA levels (DeSilva et al.).
DISEASE MODELING
· Activates peroxisome proliferator activated receptor (PPAR) co-activator 1α (PGC-1α) and prevents neurotoxicity and spatial memory impairment in rats challenged by amyloid beta (Aβ; Ashabi et al.).
This product is designed for use in the following research area(s) as part
of the highlighted workflow stage(s). Explore these workflows to learn more about the other products we
offer to support each research area.
U0126 protects cells against oxidative stress independent of its function as a MEK inhibitor.
Ong Q et al.
ACS chemical neuroscience 2015
Abstract
U0126 is a potent and selective inhibitor of MEK1 and MEK2 kinases. It has been widely used as an inhibitor for the Ras/Raf/MEK/ERK signaling pathway with over 5000 references on the NCBI PubMed database. In particular, U0126 has been used in a number of studies to show that inhibition of the Raf/MEK/ERK pathway protects neuronal cells against oxidative stress. Here, we report that U0126 can function as an antioxidant that protects PC12 cells against a number of different oxidative-stress inducers. This protective effect of U0126 is independent of its function as a MEK inhibitor, as several other MEK inhibitors failed to show similar protective effects. U0126 reduces reactive oxygen species (ROS) in cells. We further demonstrate that U0126 is a direct ROS scavenger in vitro, and the oxidation products of U0126 exhibit fluorescence. Our finding that U0126 is a strong antioxidant signals caution for its future usage as a MEK inhibitor and for interpreting some previous results.
Protein kinase C regulates human pluripotent stem cell self-renewal.
Kinehara M et al.
PloS one 2013
Abstract
BACKGROUND: The self-renewal of human pluripotent stem (hPS) cells including embryonic stem and induced pluripotent stem cells have been reported to be supported by various signal pathways. Among them, fibroblast growth factor-2 (FGF-2) appears indispensable to maintain self-renewal of hPS cells. However, downstream signaling of FGF-2 has not yet been clearly understood in hPS cells. METHODOLOGY/PRINCIPAL FINDINGS: In this study, we screened a kinase inhibitor library using a high-throughput alkaline phosphatase (ALP) activity-based assay in a minimal growth factor-defined medium to understand FGF-2-related molecular mechanisms regulating self-renewal of hPS cells. We found that in the presence of FGF-2, an inhibitor of protein kinase C (PKC), GF109203X (GFX), increased ALP activity. GFX inhibited FGF-2-induced phosphorylation of glycogen synthase kinase-3β (GSK-3β), suggesting that FGF-2 induced PKC and then PKC inhibited the activity of GSK-3β. Addition of activin A increased phosphorylation of GSK-3β and extracellular signal-regulated kinase-1/2 (ERK-1/2) synergistically with FGF-2 whereas activin A alone did not. GFX negated differentiation of hPS cells induced by the PKC activator, phorbol 12-myristate 13-acetate whereas Gö6976, a selective inhibitor of PKCα, β, and γ isoforms could not counteract the effect of PMA. Intriguingly, functional gene analysis by RNA interference revealed that the phosphorylation of GSK-3β was reduced by siRNA of PKCδ, PKCε, and ζ, the phosphorylation of ERK-1/2 was reduced by siRNA of PKCε and ζ, and the phosphorylation of AKT was reduced by PKCε in hPS cells. CONCLUSIONS/SIGNIFICANCE: Our study suggested complicated cross-talk in hPS cells that FGF-2 induced the phosphorylation of phosphatidylinositol-3 kinase (PI3K)/AKT, mitogen-activated protein kinase/ERK-1/2 kinase (MEK), PKC/ERK-1/2 kinase, and PKC/GSK-3β. Addition of GFX with a MEK inhibitor, U0126, in the presence of FGF-2 and activin A provided a long-term stable undifferentiated state of hPS cells even though hPS cells were dissociated into single cells for passage. This study untangles the cross-talk between molecular mechanisms regulating self-renewal and differentiation of hPS cells.
ERK and p38 inhibitors attenuate memory deficits and increase CREB phosphorylation and PGC-1α levels in Aβ-injected rats.
Ashabi G et al.
Behavioural brain research 2012
Abstract
In this study, we investigated the effect of intracerebroventricular administration of ERK and p38 specific inhibitors, U0126 and PD169316, respectively, on learning and memory deficits induced by amyloid beta (Aβ) in rats. To investigate the effects of these compounds on learning and memory, we performed Morris water maze (MWM) test. U0126 and/or PD169316 improved spatial learning in MWM in Aβ-injected rats, 20 days after Aβ-injection. To determine the mechanisms of action of U0126 and PD169316, we studies their effect on some intracellular signaling pathways such as Ca(+)/cAMP-response element binding protein (CREB), c-fos, and transcription factors that regulate mitochondrial biogenesis. Based on our data, CREB and c-fos levels decreased 7 days after Aβ-injection, while U0126 and/or PD169316 pretreatments significantly increased these levels. Moreover, U0126 and PD169316 activated peroxisome proliferator-activated receptor gamma coactivator-1a, nuclear respiratory factor 1, and mitochondrial transcription factor A, 7 days after Aβ-injection. Surprisingly, these factors were returned to vehicle level, 20 days after Aβ-injection. Our findings reinforce the potential neuroprotective effect of these inhibitors against the Aβ toxicity.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.