Thank you for your interest in this product.
Please provide us with your contact information and your local representative
will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
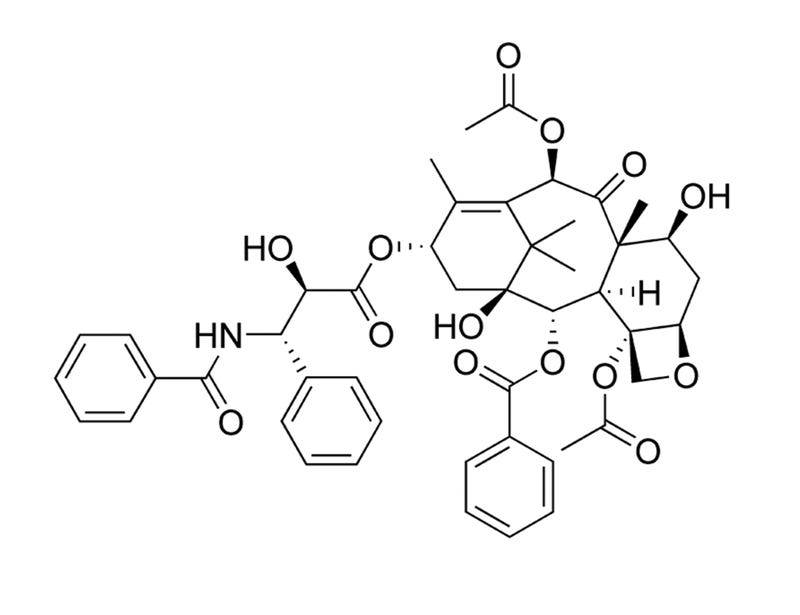
Paclitaxel is a diterpene alkaloid originally isolated from the bark of the Pacific Yew tree (Taxus brevifolia). It binds to and stabilizes microtubules, preventing their reorganization during cell division, which leads to cell cycle arrest. Paclitaxel has antitumorigenic properties and has been used as a chemotherapeutic compound (Rowinsky et al.). Many pathways have been implicated in Paclitaxel-induced apoptosis, including c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK), p38 mitogen-activated protein kinase (MAPK), and protein kinase A (PKA; Wang et al.; Reshkin et al.).
DIFFERENTIATION
· Inhibits initiation and outgrowth of neurites in vitro, through microtubule polymerization (Letourneau & Ressler).
CANCER RESEARCH
· Inhibits tumor cell growth in a variety of cancer cell lines including cervical (HeLa), lung (A549), breast (MCF-7), colon (HT-29), ovarian (OVG-1), and pancreatic (PC-Sh) carcinomas (Liebmann et al.).
· Induces abnormal multipolar spindle formation, inducing cell cycle arrest at prophase and G1 in various human cell cancer lines (Woods et al.).
· Initiates apoptosis of cancer cells through multiple mechanisms involving: p53-dependent and -independent pathways, B-cell CLL/lymphoma 2 (BCL-2) family members, cyclin-dependent kinases, p38 MAPK, PKA, and JNK/SAPK (Wang et al.; Reshkin et al.).
· Induces cyclin inhibitor p21 in MCF7 and PC3M human cancer cell lines by a mechanism dependent on the activation of RAF-1 (Blagosklonny et al.).
Cell Type
Cancer Cells and Cell Lines, Leukemia/Lymphoma Cells, Neurons
This product is designed for use in the following research area(s) as part
of the highlighted workflow stage(s). Explore these workflows to learn more about the other products we
offer to support each research area.
Paclitaxel induces apoptosis via protein kinase A- and p38 mitogen-activated protein-dependent inhibition of the Na+/H+ exchanger (NHE) NHE isoform 1 in human breast cancer cells.
Reshkin SJ et al.
Clinical cancer research : an official journal of the American Association for Cancer Research 2003
Abstract
PURPOSE: The molecular signal components essential to paclitaxel-dependent apoptosis in breast cancers are potential targets for combined therapy. However, the signal mechanisms underlying paclitaxel action still need to be better defined. EXPERIMENTAL DESIGN: In a breast cancer cell line, pharmacological agents and transient transfection with dominant interfering and constitutive active mutants were used to identify the signal transduction module involved in the regulation of paclitaxel-induced apoptosis and to evaluate its potential as a therapeutic target. RESULTS: In MDA-MB-435 cells, paclitaxel treatment stimulated the activity of both protein kinase A and p38, and inhibited the activity of the Na(+)/H(+) exchanger isoform 1 (NHE1) with similar IC(50) concentrations as for its activation of apoptosis. Activation and inhibition experiments demonstrated that protein kinase A and p38 participate sequentially upstream of the NHE1 in regulating the paclitaxel-induced apoptotic pathway. Importantly, concurrent specific inhibition of the NHE1 with paclitaxel treatment resulted in a synergistic induction of apoptosis and a reduction in the paclitaxel IC(50) for apoptosis. This sensitization of paclitaxel apoptotic action by specific inhibition of NHE1 was verified in breast cancer cell lines with different paclitaxel sensitivity. CONCLUSIONS: We have, for the first time, identified NHE1 as an essential component of paclitaxel-induced apoptosis in breast cancer cells and, importantly, identified that simultaneous inhibition of the NHE1 results in a synergistic potentiation of low-dose paclitaxel apoptotic action. As specific NHE1 inhibitors have finished Phase II/Phase III clinical trials for myocardial protection, there is the possibility for a rapid biological translation of this novel therapeutic strategy to a clinical setting.
Paclitaxel-induced cell death: where the cell cycle and apoptosis come together.
Wang TH et al.
Cancer 2000
Abstract
BACKGROUND: Compelling evidence indicates that paclitaxel kills cancer cells through the induction of apoptosis. Paclitaxel binds microtubules and causes kinetic suppression (stabilization) of microtubule dynamics. The consequent arrest of the cell cycle at mitotic phase has been considered to be the cause of paclitaxel-induced cytotoxicity. However, the biochemical events, downstream from paclitaxel's binding to microtubules, that lead to apoptosis are not well understood. METHODS: The authors examined recent scientific literature about the mechanisms by which paclitaxel exerts cytotoxicity. RESULTS: In addition to an arrest of the cell cycle at the mitotic phase in paclitaxel-treated cells, recent discoveries of activation of signaling molecules by paclitaxel and paclitaxel-induced transcriptional activation of various genes indicate that paclitaxel initiates apoptosis through multiple mechanisms. The checkpoint of mitotic spindle assembly, aberrant activation of cyclin-dependent kinases, and the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) are shown to be involved in paclitaxel-induced apoptosis. Consistent with observations that microtubules of different status (e.g., cytoskeletal microtubules vs. mitotic spindles) have different sensitivity to paclitaxel, the concentration of paclitaxel appears to be the major determinant of its apoptogenic mechanisms. CONCLUSIONS: Advances in research of the cell cycle and apoptosis have extended our understanding of the mechanisms of paclitaxel-induced cell death. Further elucidation of resistance and enhancement of paclitaxel-induced apoptosis should expedite the development of better paclitaxel-based regimens for cancer therapy.
Taxol induction of p21WAF1 and p53 requires c-raf-1.
Blagosklonny MV et al.
Cancer research 1995
Abstract
Taxol stabilizes microtubules, prevents tubulin depolymerization, and promotes tubulin bundling and is one of the most effective drugs for the treatment of metastatic breast and ovarian cancer. Although its interaction with tubulin has been well characterized, the mechanism by which taxol induces growth arrest and cytotoxicity is not well understood. Herein, we show that taxol induced dose- and time-dependent accumulation of the cyclin inhibitor p21WAF1 in both p53 wild-type and p53-null cells, although the degree of induction was greater in cells expressing wild-type p53. In MCF7 cells, wild-type p53 protein was also induced after taxol treatment, and this induction was mediated primarily by increased protein stability. Taxol induced both p21WAF1 and wild-type p53 optimally in MCF7 cells after 20-24-h exposure with an EC50(3) of 5 nM. In p53-null PC3M cells, p21WAF1 was similarly induced after 24-h exposure to taxol. Coincident with these biochemical effects, taxol altered the electrophoretic mobility of c-raf-1 and stimulated mitogen activated protein kinase. Previous depletion of c-raf-1 inhibited both the p21WAF1- and p53-inducing properties of taxol, as well as the activation of MAP kinase. These data suggest that induction of p21WAF1 by taxol requires c-raf-1 activity, but that it is not strictly dependent on wild-type p53. Furthermore, the ability of taxol to both induce wild-type p53 in MCF7 cells and activate MAP kinase is also dependent on c-raf-1 expression.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.