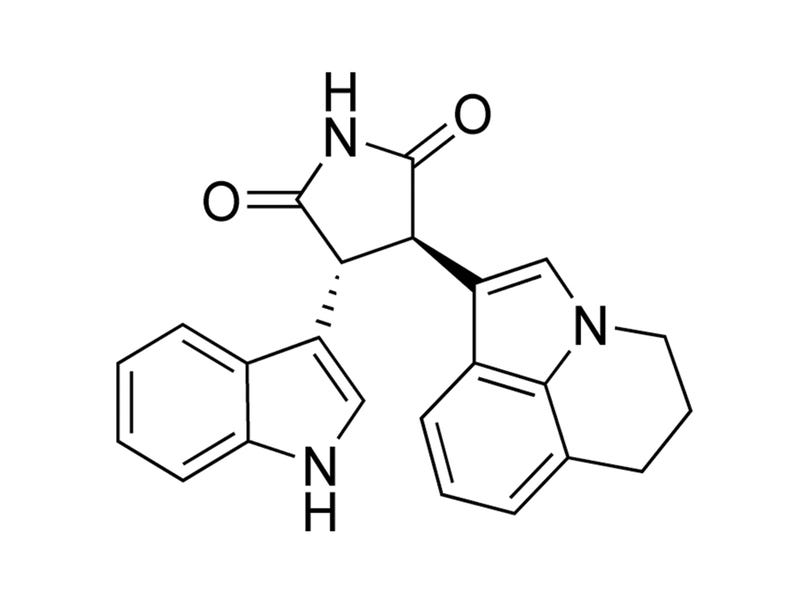
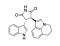
Tivantinib
HGF pathway inhibitor; Inhibits MET
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (7)
Targeting the pro-survival protein MET with tivantinib (ARQ 197) inhibits growth of multiple myeloma cells.
Neoplasia (New York, N.Y.) 2015
Abstract
The hepatocyte growth factor (HGF)/MNNG HOS transforming gene (MET) pathway regulates cell growth, survival, and migration. MET is mutated or amplified in several malignancies. In myeloma, MET is not mutated, but patients have high plasma concentrations of HGF, high levels of MET expression, and gene copy number, which are associated with poor prognosis and advanced disease. Our previous studies demonstrated that MET is critical for myeloma cell survival and its knockdown induces apoptosis. In our current study, we tested tivantinib (ARQ 197), a small-molecule pharmacological MET inhibitor. At clinically achievable concentrations, tivantinib induced apoptosis by textgreater50% in all 12 human myeloma cell lines tested. This biologic response was associated with down-regulation of MET signaling and inhibition of the mitogen-activated protein kinase and phosphoinositide 3-kinase pathways, which are downstream of the HGF/MET axis. Tivantinib was equally effective in inducing apoptosis in myeloma cell lines resistant to standard chemotherapy (melphalan, dexamethasone, bortezomib, and lenalidomide) as well as in cells that were co-cultured with a protective bone marrow microenvironment or with exogenous cytokines. Tivantinib induced apoptosis in CD138+ plasma cells from patients and demonstrated efficacy in a myeloma xenograft mouse model. On the basis of these data, we initiated a clinical trial for relapsed/refractory multiple myeloma (MM). In conclusion, MET inhibitors may be an attractive target-based strategy for the treatment of MM.
Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes ABC transporter-mediated drug resistance.
Molecular cancer therapeutics 2014
Abstract
Tivantinib (ARQ197) was first reported as a highly selective inhibitor of c-MET and is currently being investigated in a phase III clinical trial. However, as recently reported by us and another group, tivantinib showed cytotoxic activity independent of cellular c-MET status and also disrupted microtubule dynamics. To investigate if tivantinib exerts its cytotoxic activity by disrupting microtubules, we quantified polymerized tubulin in cells and xenograft tumors after tivantinib treatment. Consistent with our previous report, tivantinib reduced tubulin polymerization in cells and in mouse xenograft tumors in vivo. To determine if tivantinib directly binds to tubulin, we performed an in vitro competition assay. Tivantinib competitively inhibited colchicine but not vincristine or vinblastine binding to purified tubulin. These results imply that tivantinib directly binds to the colchicine binding site of tubulin. To predict the binding mode of tivantinib with tubulin, we performed computer simulation of the docking pose of tivantinib with tubulin using GOLD docking program. Computer simulation predicts tivantinib fitted into the colchicine binding pocket of tubulin without steric hindrance. Furthermore, tivantinib showed similar IC50 values against parental and multidrug-resistant cells. In contrast, other microtubule-targeting drugs, such as vincristine, paclitaxel, and colchicine, could not suppress the growth of cells overexpressing ABC transporters. Moreover, the expression level of ABC transporters did not correlate with the apoptosis-inducing ability of tivantinib different from other microtubule inhibitor. These results suggest that tivantinib can overcome ABC transporter-mediated multidrug-resistant tumor cells and is potentially useful against various tumors.
Tivantinib (ARQ197) Displays Cytotoxic Activity That Is Independent of Its Ability to Bind MET
Clinical Cancer Research 2013
Abstract
PURPOSE: MET, the high-affinity receptor for hepatocyte growth factor, is frequently deregulated in human cancer. Tivantinib (ARQ197; Arqule), a staurosporine derivative that binds to the dephosphorylated MET kinase in vitro, is being tested clinically as a highly selective MET inhibitor. However, the mechanism of action of tivantinib is still unclear. EXPERIMENTAL DESIGN: The activity of tivantinib was analyzed in multiple cellular models, including: cells displaying c-MET gene amplification, strictly 'addicted' to MET signaling; cells with normal c-MET gene copy number, not dependent on MET for growth; cells not expressing MET; somatic knockout cells in which the ATP-binding cleft of MET, where tivantinib binds, was deleted by homologous recombination; and a cell system 'poisoned' by MET kinase hyperactivation, where cells die unless cultured in the presence of a specific MET inhibitor. RESULTS: Tivantinib displayed cytotoxic activity independently of c-MET gene copy number and regardless of the presence or absence of MET. In both wild-type and isogenic knockout cells, tivantinib perturbed microtubule dynamics, induced G2/M arrest, and promoted apoptosis. Tivantinib did not rescue survival of cells 'poisoned' by MET kinase hyperactivation, but further incremented cell death. In all cell models analyzed, tivantinib did not inhibit HGF-dependent or -independent MET tyrosine autophosphorylation. CONCLUSIONS: We conclude that tivantinib displays cytotoxic activity via molecular mechanisms that are independent from its ability to bind MET. This notion has a relevant impact on the interpretation of clinical results, on the design of future clinical trials, and on the selection of patients receiving tivantinib treatment.
Related Products
Related Products
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
