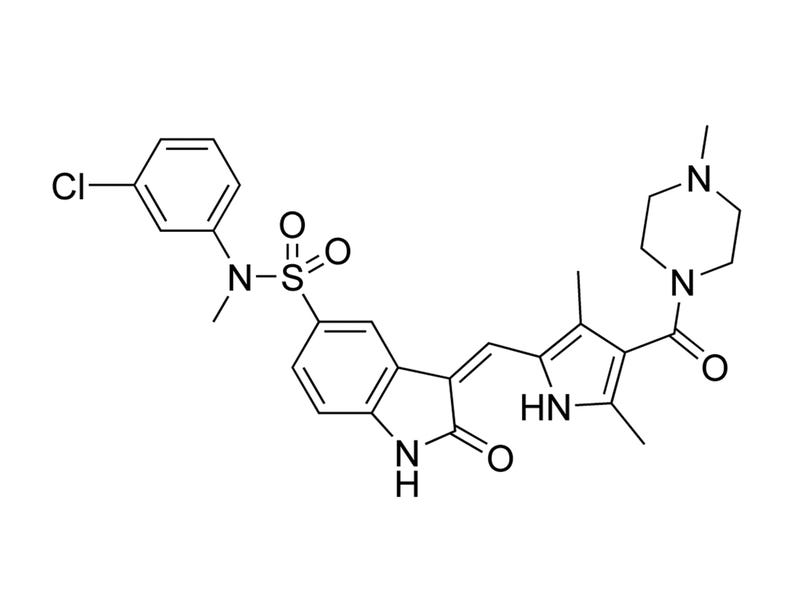
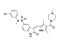
SU11274
HGF pathway inhibitor; Inhibits MET
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (4)
The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma.
Cancer research 2009
Abstract
Recurrent/metastatic head and neck cancer remains a devastating disease with insufficient treatment options. We investigated the MET receptor tyrosine kinase as a novel target for the treatment of head and neck squamous cell carcinoma (HNSCC). MET/phosphorylated MET and HGF expression was analyzed in 121 tissues (HNSCC/normal) by immunohistochemistry, and in 20 HNSCC cell lines by immunoblotting. The effects of MET inhibition using small interfering RNA/two small-molecule inhibitors (SU11274/PF-2341066) on signaling, migration, viability, and angiogenesis were determined. The complete MET gene was sequenced in 66 head and neck cancer tissue samples and eight cell lines. MET gene copy number was determined in 14 cell lines and 23 tumor tissues. Drug combinations of SU11274 with cisplatin or erlotinib were tested in SCC35/HN5 cell lines. Eighty-four percent of the HNSCC samples showed MET overexpression, whereas 18 of 20 HNSCC cell lines (90%) expressed MET. HGF overexpression was present in 45% of HNSCC. MET inhibition with SU11274/PF-2341066 abrogated MET signaling, cell viability, motility/migration in vitro, and tumor angiogenesis in vivo. Mutational analysis of 66 tumor tissues and 8 cell lines identified novel mutations in the semaphorin (T230M/E168D/N375S), juxtamembrane (T1010I/R988C), and tyrosine kinase (T1275I/V1333I) domains (incidence: 13.5%). Increased MET gene copy number was present with textgreater10 copies in 3 of 23 (13%) tumor tissues. A greater-than-additive inhibition of cell growth was observed when combining a MET inhibitor with cisplatin or erlotinib and synergy may be mediated via erbB3/AKT signaling. MET is functionally important in HNSCC with prominent overexpression, increased gene copy number, and mutations. MET inhibition abrogated MET functions, including proliferation, migration/motility, and angiogenesis. MET is a promising, novel target for HNSCC and combination approaches with cisplatin or EGFR inhibitors should be explored.
Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer.
Cancer research 2005 FEB
Abstract
Non-small cell lung cancer (NSCLC) is a difficult disease to treat. The c-Met receptor is an attractive potential target for novel therapeutic inhibition in human cancers. We provide strong evidence that c-Met is overexpressed, activated, and sometimes mutated in NSCLC cell lines and tumor tissues. Expression of c-Met was found in all (100%) of the NSCLC tumor tissues examined (n = 23) and most (89%) of the cell lines (n = 9). Sixty-one percent of tumor tissues strongly expressed total c-Met, especially adenocarcinoma (67%). Specific expression of phospho-Met (p-Met) [Y1003] and [Y1230/1234/1235] was seen by immunohistochemistry. p-Met expression was preferentially observed at the NSCLC tumor invasive fronts. c-Met alterations were identified within the semaphorin domain (E168D, L299F, S323G, and N375S) and the juxtamembrane domain (R988C, R988C + T1010I, S1058P, and alternative splice product skipping entire juxtamembrane domain) of a NSCLC cell line and adenocarcinoma tissues. We validated c-Met as potential therapeutic target using small interfering RNA down-regulation of the receptor expression by 50% to 60% in NSCLC cells. This led to inhibition of p-Met and phospho-AKT and up to 57.1 +/- 7.2% cell viability inhibition at 72 hours. The selective small molecule inhibitor of c-Met SU11274 inhibited cell viability in c-Met-expressing NSCLC cells. SU11274 also abrogated hepatocyte growth factor-induced phosphorylation of c-Met and its downstream signaling. Here, we provide first direct evidence by small interfering RNA targeting and small molecule inhibitor that c-Met is important in NSCLC biology and biochemistry. These results indicate that c-Met inhibition will be an important therapeutic strategy against NSCLC to improve its clinical outcome.
The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants.
Oncogene 2004
Abstract
Point mutations constitute a major mode of oncogenic activation of the Met receptor tyrosine kinase. Met is aberrantly activated in many types of human malignancies and its deregulated activity is correlated with aggressive tumor traits such as abnormal proliferation and survival, leading to tumor growth, local invasion and metastasis. Here we report that the Met kinase inhibitor SU11274 differentially affects the kinase activity and subsequent signaling of various mutant forms of Met. Two Met variants tested, M1268T and H1112Y, were potently inhibited by 2 microM SU11274, while two other variants, L1213V and Y1248H, remained resistant under similar experimental conditions. Inhibition of the kinase altered cell proliferation, morphology and motility, while cells containing resistant mutants appeared unaffected by the compound. The basis for the sensitivity or resistance to SU11274 is discussed in terms of the position of the mutations predicted from a homology model.
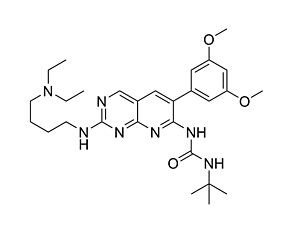
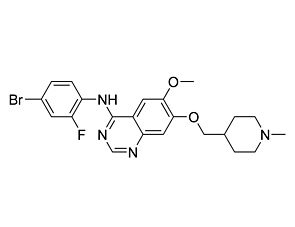
Related Products
Related Products
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
