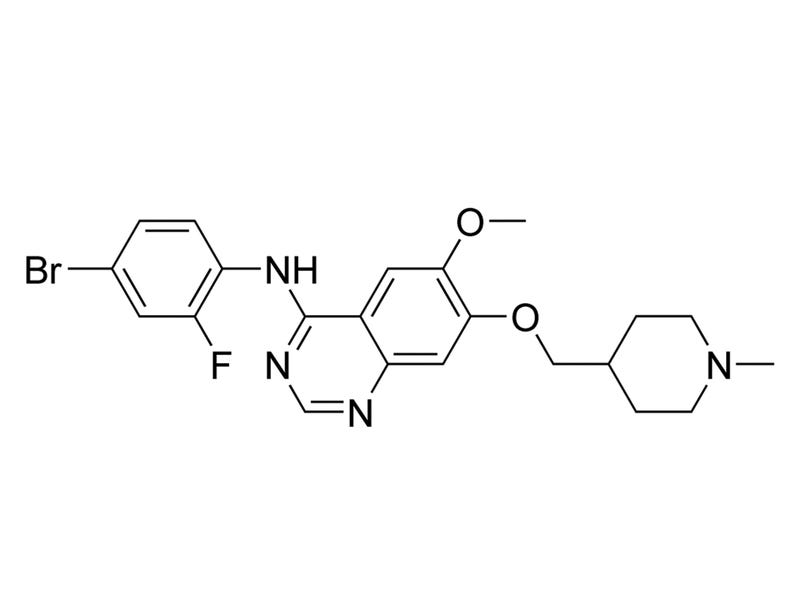
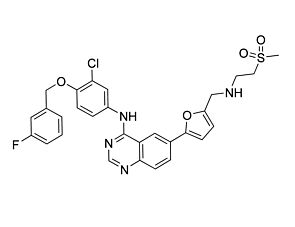
Vandetanib
Tyrosine kinase inhibitor; Inhibits VEGFR1, KDR, FLT-4, EGFR, FGFR, ABL, RET, and SRC
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (6)
Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain.
PloS one 2012
Abstract
Chronic myeloid leukemia (CML) is caused by the kinase activity of the BCR-Abl fusion protein. The Abl inhibitors imatinib, nilotinib and dasatinib are currently used to treat CML, but resistance to these inhibitors is a significant clinical problem. The kinase inhibitor bosutinib has shown efficacy in clinical trials for imatinib-resistant CML, but its binding mode is unknown. We present the 2.4 Å structure of bosutinib bound to the kinase domain of Abl, which explains the inhibitor's activity against several imatinib-resistant mutants, and reveals that similar inhibitors that lack a nitrile moiety could be effective against the common T315I mutant. We also report that two distinct chemical compounds are currently being sold under the name bosutinib"�
Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: current status and future directions.
The oncologist 2009
Abstract
Vandetanib is a novel, orally available inhibitor of different intracellular signaling pathways involved in tumor growth, progression, and angiogenesis: vascular endothelial growth factor receptor-2, epidermal growth factor receptor, and REarranged during Transfection tyrosine kinase activity. Phase I clinical trials have shown that vandetanib is well tolerated as a single agent at daily doses textless or =300 mg. In the phase II setting, negative results were observed with vandetanib in small cell lung cancer, metastatic breast cancer, and multiple myeloma. In contrast, three randomized phase II studies showed that vandetanib prolonged the progression-free survival (PFS) time of patients with non-small cell lung cancer (NSCLC) as a single agent when compared with gefitinib or when added to chemotherapy. Rash, diarrhea, hypertension, fatigue, and asymptomatic QTc prolongation were the most common adverse events. Antitumor activity was also observed in medullary thyroid cancer. Four randomized phase III clinical trials in NSCLC are exploring the efficacy of vandetanib in combination with docetaxel, the Zactima in cOmbination with Docetaxel In non-small cell lung Cancer (ZODIAC) trial, or with pemetrexed, the Zactima Efficacy with Alimta in Lung cancer (ZEAL) trial, or as a single agent, the Zactima Efficacy when Studied versus Tarceva (ZEST) and the Zactima Efficacy trial for NSCLC Patients with History of EGFR-TKI chemo-Resistance (ZEPHYR) trials. Based on a press release by the sponsor of these trials, the PFS time was longer with vandetanib in the ZODIAC and ZEAL trials; the ZEST trial was negative for its primary superiority analysis, but was successful according to a preplanned noninferiority analysis of PFS. Ongoing phase II and III clinical trials will better define the appropriate schedule, the optimal setting of evaluation, and the safety of long-term use of vandetanib.
1H-1,2,4-triazol-3-yl-anilines: novel potent inhibitors of vascular endothelial growth factor receptors 1 and 2.
Chemical biology & drug design 2007
Abstract
Novel derivatives of 1,2,4-triazoles are described as potent ATP-competitive inhibitors of vascular endothelial growth factor receptors I and II (VEGFR-1/2). A number of compounds display VEGFR-2 inhibitory activity comparable to that of Vatalanib and Vandetanib in both homogenous time-resolved fluorescence enzymatic and cellular assays. Several active molecules feature high intrinsic permeability (textgreater30 x 10(-5) cm/min) across Caco-2 cell monolayer.
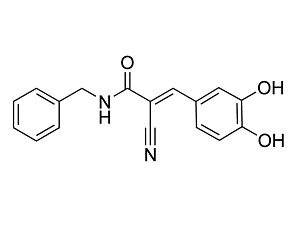
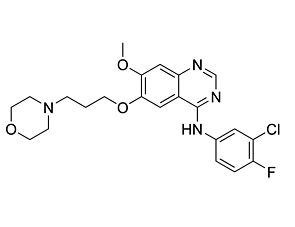
Related Products
Related Products
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
