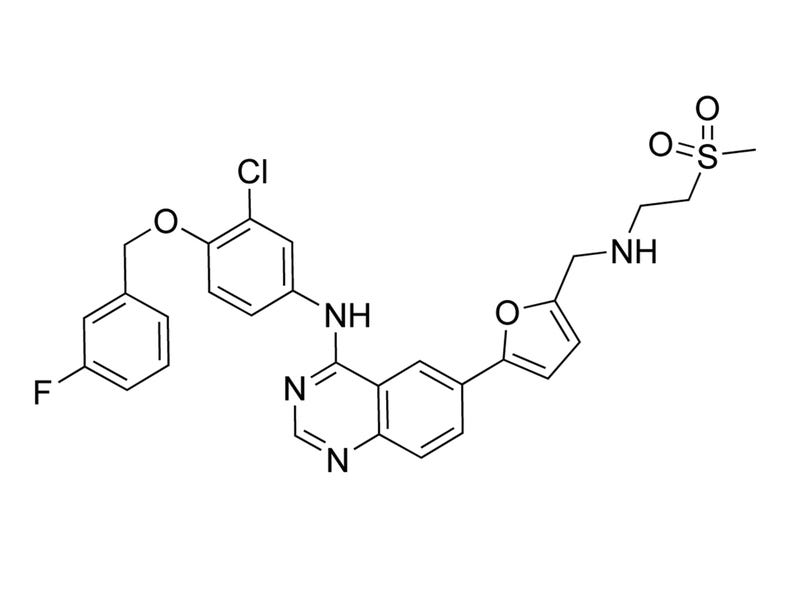
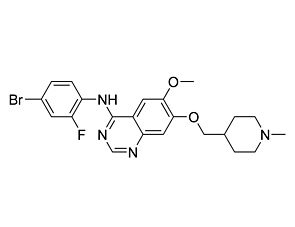
Lapatinib
Tyrosine kinase inhibitor; Inhibits EGFR and HER2
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (3)
A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells.
Cancer research 2004
Abstract
GW572016 (Lapatinib) is a tyrosine kinase inhibitor in clinical development for cancer that is a potent dual inhibitor of epidermal growth factor receptor (EGFR, ErbB-1) and ErbB-2. We determined the crystal structure of EGFR bound to GW572016. The compound is bound to an inactive-like conformation of EGFR that is very different from the active-like structure bound by the selective EGFR inhibitor OSI-774 (Tarceva) described previously. Surprisingly, we found that GW572016 has a very slow off-rate from the purified intracellular domains of EGFR and ErbB-2 compared with OSI-774 and another EGFR selective inhibitor, ZD-1839 (Iressa). Treatment of tumor cells with these inhibitors results in down-regulation of receptor tyrosine phosphorylation. We evaluated the duration of the drug effect after washing away free compound and found that the rate of recovery of receptor phosphorylation in the tumor cells reflected the inhibitor off-rate from the purified intracellular domain. The slow off-rate of GW572016 correlates with a prolonged down-regulation of receptor tyrosine phosphorylation in tumor cells. The differences in the off-rates of these drugs and the ability of GW572016 to inhibit ErbB-2 can be explained by the enzyme-inhibitor structures.
Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways.
Oncogene 2002
Abstract
Dual EGFR/erbB2 inhibition is an attractive therapeutic strategy for epithelial tumors, as ligand-induced erbB2/EGFR heterodimerization triggers potent proliferative and survival signals. Here we show that a small molecule, GW572016, potently inhibits both EGFR and erbB2 tyrosine kinases leading to growth arrest and/or apoptosis in EGFR and erbB2-dependent tumor cell lines. GW572016 markedly reduced tyrosine phosphorylation of EGFR and erbB2, and inhibited activation of Erk1/2 and AKT, downstream effectors of proliferation and cell survival, respectively. Complete inhibition of activated AKT in erbB2 overexpressing cells correlated with a 23-fold increase in apoptosis compared with vehicle controls. EGF, often elevated in cancer patients, did not reverse the inhibitory effects of GW572016. These observations were reproduced in vivo, where GW572016 treatment inhibited activation of EGFR, erbB2, Erk1/2 and AKT in human tumor xenografts. Erk1/2 and AKT represent potential biomarkers to assess the clinical activity of GW572016. Inhibition of activated AKT in EGFR or erbB2-dependent tumors by GW572016 may lead to tumor regressions when used as a monotherapy, or may enhance the anti-tumor activity of chemotherapeutics, since constitutive activation of AKT has been linked to chemo-resistance.
The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo.
Molecular cancer therapeutics 2001 DEC
Abstract
The epidermal growth factor receptor (EGFR) and ErbB-2 transmembrane tyrosine kinases are currently being targeted by various mechanisms in the treatment of cancer. GW2016 is a potent inhibitor of the ErbB-2 and EGFR tyrosine kinase domains with IC50 values against purified EGFR and ErbB-2 of 10.2 and 9.8 nM, respectively. This report describes the efficacy in cell growth assays of GW2016 on human tumor cell lines overexpressing either EGFR or ErbB-2: HN5 (head and neck), A-431 (vulva), BT474 (breast), CaLu-3 (lung), and N87 (gastric). Normal human foreskin fibroblasts, nontumorigenic epithelial cells (HB4a), and nonoverexpressing tumor cells (MCF-7 and T47D) were tested as negative controls. After 3 days of compound exposure, average IC50 values for growth inhibition in the EGFR- and ErbB-2-overexpressing tumor cell lines were textless 0.16 microM. The average selectivity for the tumor cells versus the human foreskin fibroblast cell line was 100-fold. Inhibition of EGFR and ErbB-2 receptor autophosphorylation and phosphorylation of the downstream modulator, AKT, was verified by Western blot analysis in the BT474 and HN5 cell lines. As a measure of cytotoxicity versus growth arrest, the HN5 and BT474 cells were assessed in an outgrowth assay after a transient exposure to GW2016. The cells were treated for 3 days in five concentrations of GW2016, and cell growth was monitored for an additional 12 days after removal of the compound. In each of these tumor cell lines, concentrations of GW2016 were reached where outgrowth did not occur. Furthermore, growth arrest and cell death were observed in parallel experiments, as determined by bromodeoxyuridine incorporation and propidium iodide staining. GW2016 treatment inhibited tumor xenograft growth of the HN5 and BT474 cells in a dose-responsive manner at 30 and 100 mg/kg orally, twice daily, with complete inhibition of tumor growth at the higher dose. Together, these results indicate that GW2016 achieves excellent potency on tumor cells with selectivity for tumor versus normal cells and suggest that GW2016 has value as a therapy for patients with tumors overexpressing either EGFR or ErbB-2.
Related Products
Related Products
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
