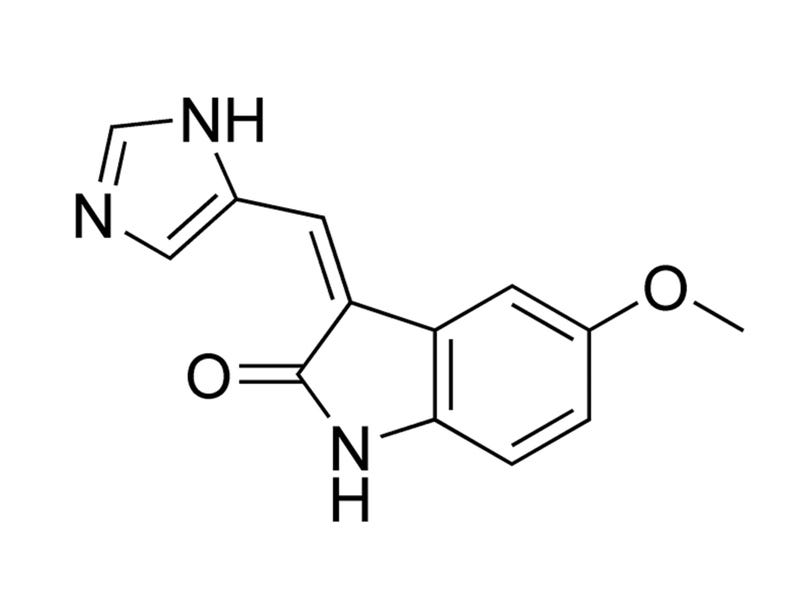
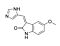
SU9516
Cyclin/CDK pathway inhibitor; Inhibits CDK1 and CDK2
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (5)
Inhibition of Cdk2 kinase activity selectively targets the CD44�?�/CD24�?�/Low stem-like subpopulation and restores chemosensitivity of SUM149PT triple-negative breast cancer cells.
International journal of oncology 2014
Abstract
Inflammatory breast cancer (IBC) is an angioinvasive and most aggressive type of advanced breast cancer characterized by rapid proliferation, chemoresistance, early metastatic development and poor prognosis. IBC tumors display a triple-negative breast cancer (TNBC) phenotype characterized by centrosome amplification, high grade of chromosomal instability (CIN) and low levels of expression of estrogen receptor α (ERα), progesterone receptor (PR) and HER-2 tyrosine kinase receptor. Since the TNBC cells lack these receptors necessary to promote tumor growth, common treatments such as endocrine therapy and molecular targeting of HER-2 receptor are ineffective for this subtype of breast cancer. To date, not a single targeted therapy has been approved for non-inflammatory and inflammatory TNBC tumors and combination of conventional cytotoxic chemotherapeutic agents remains the standard therapy. IBC tumors generally display activation of epithelial to mesenchymal transition (EMT) that is functionally linked to a CD44+/CD24-/Low stem-like phenotype. Development of EMT and consequent activation of stemness programming is responsible for invasion, tumor self-renewal and drug resistance leading to breast cancer progression, distant metastases and poor prognosis. In this study, we employed the luminal ER+ MCF-7 and the IBC SUM149PT breast cancer cell lines to establish the extent to which high grade of CIN and chemoresistance were mechanistically linked to the enrichment of CD44+/CD24low/- CSCs. Here, we demonstrate that SUM149PT cells displayed higher CIN than MCF-7 cells characterized by higher percentage of structural and numerical chromosomal aberrations. Moreover, centrosome amplification, cyclin E overexpression and phosphorylation of retinoblastoma (Rb) were restricted to the stem-like CD44+/CD24-/Low subpopulation isolated from SUM149PT cells. Significantly, CD44+/CD24-/Low CSCs displayed resistance to conventional chemotherapy but higher sensitivity to SU9516, a specific cyclin-dependent kinase 2 (Cdk2) inhibitor, demonstrating that aberrant activation of cyclin E/Cdk2 oncogenic signaling is essential for the maintenance and expansion of CD44+/CD24-/Low CSC subpopulation in IBC. In conclusion, our findings propose a novel therapeutic approach to restore chemosensitivity and delay recurrence of IBC tumors based on the combination of conventional chemotherapy with small molecule inhibitors of the Cdk2 cell cycle kinase.
The three-substituted indolinone cyclin-dependent kinase 2 inhibitor 3-[1-(3H-imidazol-4-yl)-meth-(Z)-ylidene]-5-methoxy-1,3-dihydro-indol-2-one (SU9516) kills human leukemia cells via down-regulation of Mcl-1 through a transcriptional mechanism.
Molecular pharmacology 2006
Abstract
Mechanisms of lethality of the three-substituted indolinone and putatively selective cyclin-dependent kinase (CDK)2 inhibitor 3-[1-(3H-imidazol-4-yl)-meth-(Z)-ylidene]-5-methoxy-1,3-dihydro-indol-2-one (SU9516) were examined in human leukemia cells. Exposure of U937 and other leukemia cells to SU9516 concentrations textgreater or =5 microM rapidly (i.e., within 4 h) induced cytochrome c release, Bax mitochondrial translocation, and apoptosis in association with pronounced down-regulation of the antiapoptotic protein Mcl-1. These effects were associated with inhibition of phosphorylation of the carboxyl-terminal domain (CTD) of RNA polymerase (Pol) II on serine 2 but not serine 5. Reverse transcription-polymerase chain reaction analysis revealed pronounced down-regulation of Mcl-1 mRNA levels in SU9516-treated cells. Similar results were obtained in Jurkat and HL-60 leukemia cells. Furthermore, cotreatment with the proteasome inhibitor N-benzoyloxycarbonyl (Z)-Leu-Leu-leucinal (MG132) blocked SU9516-mediated Mcl-1 down-regulation, implicating proteasomal degradation in diminished expression of this protein. Ectopic expression of Mcl-1 largely blocked SU9516-induced cytochrome c release, Bax translocation, and apoptosis, whereas knockdown of Mcl-1 by small interfering RNA potentiated SU9516 lethality, confirming the functional contribution of Mcl-1 down-regulation to SU9516-induced cell death. It is noteworthy that SU9516 treatment resulted in a marked increase in reactive oxygen species production, which was diminished, along with cell death, by the free radical scavenger N-acetylcysteine (NAC). We were surprised to find that NAC blocked SU9516-mediated inhibition of RNA Pol II CTD phosphorylation on serine 2, reductions in Mcl-1 mRNA levels, and Mcl-1 down-regulation. Together, these findings suggest that SU9516 kills leukemic cells through inhibition of RNA Pol II CTD phosphorylation in association with oxidative damage and down-regulation of Mcl-1 at the transcriptional level, culminating in mitochondrial injury and cell death.
SU9516: biochemical analysis of cdk inhibition and crystal structure in complex with cdk2.
Biochemical and biophysical research communications 2003
Abstract
SU9516 is a 3-substituted indolinone compound with demonstrated potent and selective inhibition toward cyclin dependent kinases (cdks). Here, we describe the kinetic characterization of this inhibition with respect to cdk2, 1, and 4, along with the crystal structure in complex with cdk2. The molecule is competitive with respect to ATP for cdk2/cyclin A, with a K(i) value of 0.031 microM. Similarly, SU9516 inhibits cdk2/cyclin E and cdk1/cyclin B1 in an ATP-competitive manner, although at a 2- to 8-fold reduced potency. In contrast, the compound exhibited non-competitive inhibition with respect to ATP toward cdk4/cyclin D1, with a 45-fold reduced potency. The X-ray crystal structure of SU9516 bound to cdk2 revealed interactions between the molecule and Leu83 and Glu81 of the kinase. This study should aid in the development of more potent and selective cdk inhibitors for potential therapeutic agents.
Related Products
Related Products

Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
