Make more informed purchasing decisions with our new product availability and delivery estimate feature, now available on all product pages, in your cart, and during checkout.
Sign In
New to STEMCELL?
Register for an account to quickly and easily purchase products online and for one-click access to all educational content.
Thank you for your interest in this product.
Please provide us with your contact information and your local representative
will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
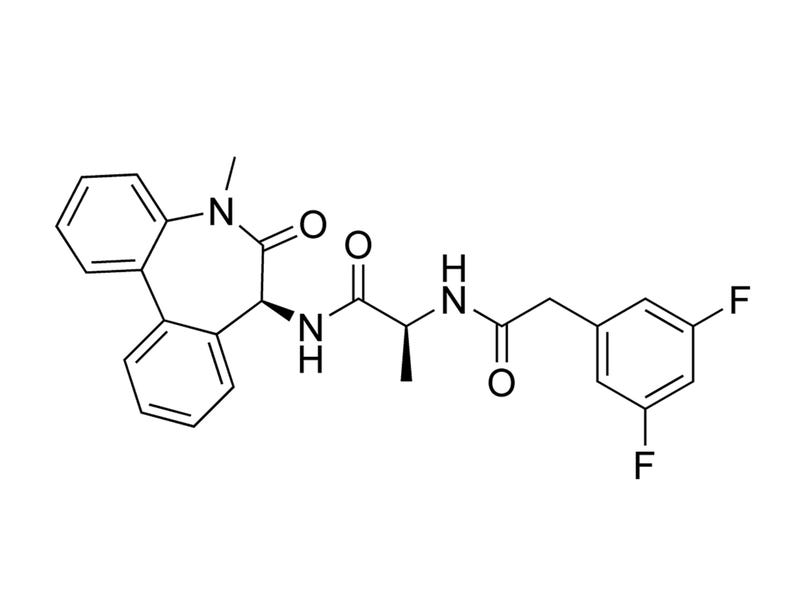
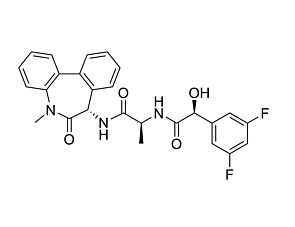
DBZ is a diazepine inhibitor of γ-secretase, which cleaves transmembrane proteins including Notch, amyloid precursor protein (APP), and Ephrin-B2 (Borgegard et al). DBZ blocks the cleavage of Notch into its active signalling effector, Notch intracellular domain, with an IC₅₀ of 1.7 nM (Milano et al.).
REPROGRAMMING
· Enables reprogramming of human keratinocytes to induced pluripotent stem cells in the absence of oncogenic reprogramming factors KLF4 and c-MYC (Ichida et al.).
DIFFERENTIATION
· Induces intestinal cell apoptosis & goblet cell metaplasia in rats; attenuates the reduction of paneth cells and goblet cells caused by tuberous sclerosis 2 (TSC2) inhibition (Milano et al.; Zhou et al.).
METABOLISM
· Improves glucose homeostasis and mediates a metabolic shift toward the utilization of fat as the energy source in mice (Bi et al.).
CANCER RESEARCH
· Induces differentiation of intestinal adenomas in Apc(Min) transgenic mice (van Es et al.).
· Decreases the production of inflammatory cytokines by alloreactive T cells after bone marrow transplantation in mice, reducing the severity of graft-versus-host disease (Tran et al.).
Cell Type
Cancer Cells and Cell Lines, Intestinal Cells, Pluripotent Stem Cells, T Cells
This product is designed for use in the following research area(s) as part
of the highlighted workflow stage(s). Explore these workflows to learn more about the other products we
offer to support each research area.
TSC2/mTORC1 signaling controls Paneth and goblet cell differentiation in the intestinal epithelium.
Zhou Y et al.
Cell death & disease 2015 JAN
Abstract
The intestinal mucosa undergoes a continual process of proliferation, differentiation and apoptosis, which is regulated by multiple signaling pathways. Notch signaling is critical for the control of intestinal stem cell maintenance and differentiation. However, the precise mechanisms involved in the regulation of differentiation are not fully understood. Previously, we have shown that tuberous sclerosis 2 (TSC2) positively regulates the expression of the goblet cell differentiation marker, MUC2, in intestinal cells. Using transgenic mice constitutively expressing a dominant negative TSC2 allele, we observed that TSC2 inactivation increased mTORC1 and Notch activities, and altered differentiation throughout the intestinal epithelium, with a marked decrease in the goblet and Paneth cell lineages. Conversely, treatment of mice with either Notch inhibitor dibenzazepine (DBZ) or mTORC1 inhibitor rapamycin significantly attenuated the reduction of goblet and Paneth cells. Accordingly, knockdown of TSC2 activated, whereas knockdown of mTOR or treatment with rapamycin decreased, the activity of Notch signaling in the intestinal cell line LS174T. Importantly, our findings demonstrate that TSC2/mTORC1 signaling contributes to the maintenance of intestinal epithelium homeostasis by regulating Notch activity.
Notch inhibition allows oncogene-independent generation of iPS cells.
Ichida JK et al.
Nature chemical biology 2014 AUG
Abstract
The reprogramming of somatic cells to pluripotency using defined transcription factors holds great promise for biomedicine. However, human reprogramming remains inefficient and relies either on the use of the potentially dangerous oncogenes KLF4 and CMYC or the genetic inhibition of the tumor suppressor gene p53. We hypothesized that inhibition of signal transduction pathways that promote differentiation of the target somatic cells during development might relieve the requirement for non-core pluripotency factors during induced pluripotent stem cell (iPSC) reprogramming. Here, we show that inhibition of Notch greatly improves the efficiency of iPSC generation from mouse and human keratinocytes by suppressing p21 in a p53-independent manner and thereby enriching for undifferentiated cells capable of long-term self-renewal. Pharmacological inhibition of Notch enabled routine production of human iPSCs without KLF4 and CMYC while leaving p53 activity intact. Thus, restricting the development of somatic cells by altering intercellular communication enables the production of safer human iPSCs.
Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity.
Bi P et al.
Nature medicine 2014 AUG
Abstract
Beige adipocytes in white adipose tissue (WAT) are similar to classical brown adipocytes in that they can burn lipids to produce heat. Thus, an increase in beige adipocyte content in WAT browning would raise energy expenditure and reduce adiposity. Here we report that adipose-specific inactivation of Notch1 or its signaling mediator Rbpj in mice results in browning of WAT and elevated expression of uncoupling protein 1 (Ucp1), a key regulator of thermogenesis. Consequently, as compared to wild-type mice, Notch mutants exhibit elevated energy expenditure, better glucose tolerance and improved insulin sensitivity and are more resistant to high fat diet-induced obesity. By contrast, adipose-specific activation of Notch1 leads to the opposite phenotypes. At the molecular level, constitutive activation of Notch signaling inhibits, whereas Notch inhibition induces, Ppargc1a and Prdm16 transcription in white adipocytes. Notably, pharmacological inhibition of Notch signaling in obese mice ameliorates obesity, reduces blood glucose and increases Ucp1 expression in white fat. Therefore, Notch signaling may be therapeutically targeted to treat obesity and type 2 diabetes.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.