Optimized Workflows for High-Efficiency Genome Editing in Stem and Primary Cell Types
CRISPR-Cas genome editing in cell culture systems is a powerful technique for disease modeling and the development of cellular therapies. Compared to work with immortalized cell lines, genome editing of stem and primary cells presents unique challenges, including issues related to efficient delivery and expression of CRISPR machinery, clonogenicity, and cytotoxicity. In this webinar, our in-house expert, Dr. Ashley Watson, discusses optimized workflows for CRISPR-Cas9 genome editing in human pluripotent stem cells and primary T cells.
CRISPR-Cas genome editing in cell culture systems is a powerful technique for disease modeling and the development of cellular therapies. Compared to work with immortalized cell lines, genome editing of stem and primary cells presents unique challenges, including issues related to efficient delivery and expression of CRISPR machinery, clonogenicity, and cytotoxicity. In this webinar, our in-house expert, Dr. Ashley Watson, discusses optimized workflows for CRISPR-Cas9 genome editing in human pluripotent stem cells and primary T cells.
Webinar Q&A
Below is a list of the questions submitted by participants during the live viewing of the webinar which could not be answered due to the time restriction. Media System 1. How does mTeSR™ Plus compared to TeSR™-E8™ for stem cell maintenance? mTeSR™ Plus is a stabilized version of the most-published mTeSR™1 formulation, whereas TeSR™-E8™ is a highly defined minimal formula based on the E8™ formulation, containing only the 8 most critical components required for hPSC maintenance. Both can be used to maintain consistent pluripotent stem cell cultures. We recommend using mTeSR™ Plus for routine, general hPSC maintenance, and TeSR™-E8™ for applications that require animal-component free or low protein content culture environments. 2. How often do we need to change the medium when culturing hPSCs with mTeSR™ Plus compared to mTeSR™1? mTeSR™ Plus is a stabilized version of mTeSR™1. As such, feeding schedules are much more flexible with this medium. To skip a single day (48 hours), we recommend performing a “single feed” as usual (for example, 2 mL per well of a 6-well plate). To skip two consecutive days (72 hours), perform a “double feed” (for example, 4 mL per well of a 6-well plate). Of course, cultures can still be fed every day with mTeSR™ Plus—it’s all up to your desired schedule. To see some examples of feeding schedules, please refer to page 7 of our mTeSR Plus Information Booklet. 3. When to deliver RNP complex, are iPSCs clonal? how to generate clonal iPSC? The schematic below outlines our hPSC genome editing workflow. When using physical (electroporation) transfection methods, we recommend to deliver the RNP complex into a single-cell suspension of hPSCs and plate those cells at a relatively high density after delivery. For chemical transfection methods, we recommend to plate hPSCs as a single-cell suspension 16 - 24 hours prior to RNP delivery. After delivery, we recommend to culture the cells for 48 - 96 hours to allow genome editing to occur. A mixed population of cells harboring different edits will be present in the culture, and editing efficiency can be monitored using the ArciTect™ T7 Endonuclease I Kit, Sanger sequencing of the target site followed by software-based analysis (1), or for additional strategies refer to the Evaluation of Genome Editing (2). Alternatively, individual clones can be derived from the mixed population of cells using single-cell fluorescence-activated cell sorting (FACS) or by plating the cells at clonal density. 4. Do you have any recommendations for generating clonal edited cell lines? A major challenge for CRISPR experiments in cultured cells lies in the generation of clonal cell lines due to low survival rates after genome editing procedures and loss of proliferative capacity. As the efficiency of a given editing event is never 100%, genetically mixed cell populations are generated after editing. In these cases, one can dissociate single cells and replate at very low density to clone a genetically homogeneous population of cells. However, this approach is only applicable to cell types that are amenable to cloning. Optimization of pre- and post-editing media culture conditions can help minimize experimental variability and provide a permissive culture environment to support generation of functional and viable edited cell clones. 5. Can CloneR™ increase single cell survival rate after sorting? At which time point should I add CloneR™ into the medium? Should I add it everyday after single cell sorting? CloneR™ is a serum-free supplement designed to increase the cloning efficiency and single-cell survival of human embryonic stem cells (ES cells) and induced pluripotent stem cells (iPS cells). CloneR™ enables the robust generation of clonal cell lines without single-cell adaptation, thus minimizing the risk of acquiring genetic abnormalities. When single-cell sorting, we recommend to sort cells directly into TeSR™ medium supplemented with CloneR™ (cloning medium) and culture the cells for 2 days without a medium change. On day 3, we recommend to perform a complete medium change with cloning medium. On the fourth day, we recommend to add cloning medium at 25% initial seed volume. We then recommend daily medium changes with hPSC maintenance medium (i.e. mTeSR™1, mTeSR™ Plus, or TeSR™-E8™ without CloneR™) until colonies are ready to be picked. For complete instructions on thawing, preparation, storage, and usage of CloneR™, refer to the Product Information Sheet (Document #DX21725), available at www.stemcell.com or contact us to request a copy. 5. Where exactly is the cut-site for a particular guide RNA? Cas9 will generate a break site 3 - 4 nucleotides upstream of the PAM sequence. 6. What strategies exist to enhance HDR rate in hPSCs? Significant efforts have been made to optimize methods for HDR-dependent knock-in editing in many different cell types (3). To overcome the low efficiency of HDR, some groups have used small molecule inhibitors and/or activators to promote HDR and suppress NHEJ (4 - 9), or to synchronize the cell cycle (10, 11). More recently, physical coupling of the donor DNA template and CRISPR-Cas machinery demonstrated increased HDR frequencies (12 - 15) that bypass the possible toxicity associated with pharmacological manipulation. Alternatively, others have opted to enrich for HDR-edited cells through positive/negative selection of marker genes (16 - 18). An example of this type of strategy was recently employed in HSPCs using RNP-based delivery of CRISPR-Cas9 coupled with packing of the donor DNA template in AAV6 to accommodate a GFP reporter gene (16). This enabled enrichment of precisely edited cells by fluorescence-activated cell sorting (FACS). A similar strategy utilizing donor DNA encoded by AAV6 was employed in human PSCs to enable efficient precise genome editing (19). 7. Is it possible to synchronize hPSCs before transfection to enrich for cells in S/G2 phase? It is generally quite challenging to synchronize stem and primary cell types, and at STEMCELL we have not attempted to synchronize cells prior to genome editing. However, a recent study (20) screened inhibitors of cell cycle progression in hPSCS. They identified that low doses of the microtubule polymerization inhibitor nocodazole as the most efficient small molecule to synchronize hPSCs in G2/M phase without affecting pluripotency or karyotypic stability. Since homology-dependent repair (HDR), which is necessary for knock-in genome editing, is most efficient during post-replicative phases of the cell cycle i.e. G2/M, synchronization of hPSCs in G2/M may improve knock-in editing efficiency (10, 11). Caution should be taken with pharmacological manipulation of sensitive cell types, such as hPSCs, since it could negatively impact fundamental cell characteristics, genetic stability, and the overall quality of the cells. 8. Generation of iPSCs often utilizes p53 inhibition to enhance reprogramming. Has this strategy been used to enhance CRISPR-Cas9 genome editing efficiency? As genome editing typically relies on induction of a DNA DSB, in most cell types including hPSCs the p53-dependent DNA damage response will become activated during the process to induce cell cycle arrest or apoptosis (21, 22). Therefore, inhibition of p53 can function to enhance editing efficiency (23) and some protocols recommend treatment with p53 inhibitors (24). As with small molecule modifiers of DNA repair and/or the cell cycle, caution should be taken with this type of pharmacological manipulation of sensitive cells, such as hPSCs, since it could negatively impact fundamental cell characteristics, genetic stability, and the overall quality of the cells. 9. What are off-target effects of CRISPR-Cas9 and what strategies exist to monitor off-target genome editing? Genome editing may induce off-target effects that are both sequence-dependent and -independent, including activation of the immune and/or DNA damage response, as well as mutation(s) at unintended locations in the genome. Cas9 can tolerate ~1–2 bp mismatches in gRNA-DNA sequence (25). Along the length of gRNA, PAM-proximal mismatches have been observed to be less well tolerated than PAM-distal mismatches (26, 27). Most gRNA design tools account for this when ranking potential gRNAs for a given target; however, it is important to assess potential sequence-dependent off-target effects using unbiased or targeted genomic assays to ensure genome editing specificity. Sequence-independent off-target effects are more challenging to identify but have the potential to negatively impact cell viability and function. To rule out off-target mutation events caused by CRISPR-Cas in gene-edited cells, PCR amplification and sequencing of potential off-target sites predicted by gRNA design programs represents the most accessible method to identify the presence of off-target edits. More systematic and unbiased methods utilizing genome-wide sequencing to detect potential off-target mutations are available, including genome-wide unbiased identification of double-stranded breaks enabled by sequencing (GUIDE-seq) (28), high-throughput, genome-wide, translocation sequencing (HTGTS) (29), and DISCOVER-seq (30). Whenever possible, it is advised to generate multiple independent clones from one or multiple rounds of gene targeting for further characterization. Alternatively, multiple gRNA designs can be used to target the same gene in parallel experiments to rule out the possibility of off-target edits leading to a particular phenotype. 10. How do you deliver RNP into T cells and hPSCs? How much RNP should I add? Is cell density an important factor during transfection and/or in pre- and post-editing culture? We routinely deliver the CRISPR-Cas9 RNP into T cells using electroporation, and into hPSCs using either electroporation or chemical transfection approaches. It is important to maintain a certain cell density for transfection and during pre- and post-editing culture, and there exists a balance between transfection efficiency, cell survival, and cell expansion. Our Technical Bulletins contain step-by-step instructions for pre- and post-editing culture conditions including cell density, RNP preparation, and delivery of the RNP complex in hPSCs (31) or human primary T cells (32). 11. Is electroporation the only way to deliver RNP into hPSCs? It is also possible to deliver CRISPR-Cas9 RNP complexes into hPSCs using chemical-based transfection methods. For step-by-step instructions regarding RNP delivery using the Mirus® TransIT-X2® dynamic delivery system, refer to our Technical Bulletin: Genome Editing of Human Pluripotent Stem Cells (31), available at www.stemcell.com or contact us to request a copy.- Sentmanat MF et al. (2018) A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci Rep 8(1): 888.
- STEMCELL Technologies (2018) Evaluation of Genome Editing (Document #27126).
- Liu M et al. (2019) Methodologies for Improving HDR Efficiency. Front Genet 9: 691.
- Chu VT et al. (2015) Increasing the Efficiency of Homology-Directed Repair for CRISPR-Cas9-induced Precise Gene Editing in Mammalian Cells. Nat Biotech 33: 543-548.
- Yu C et al. (2015) Small Molecules Enhance CRISPR Genome Editing in Pluripotent Stem Cells. Cell Stem Cell 16(2):142-147.
- Robert F et al. (2015) Pharmacological Inhibition of DNA-PK Stimulates Cas9-mediated Genome Editing. Genome Med 7: 93.
- Maruyama T et al. (2015) Increasing the Efficiency of Precise Genome Editing with CRISPR-Cas9 by Inhibition of Nonhomologous End Joining. Nat Biotechnol 33(5): 538-542.
- Pinder J et al. (2015) Nuclear Domain ‘Knock-in’ Screen for the Evaluation and Identification of Small Molecule Enhancers of CRISPR-based Genome Editing. Nucleic Acids Res 43(19): 9379-9392.
- Riesenberg S and Maricic T (2018) Targeting Repair Pathways with Small Molecules Increases Precise Genome Editing in Pluripotent Stem Cells. Nat Commun 9(1): 2164.
- Lin S et al. (2014) Enhanced Homology-directed Human Genome Engineering by Controlled Timing of CRISPR/Cas9 Delivery. Elife 3: e04766.
- Yang D et al. (2016) Enrichment of G2/M Cell Cycle Phase in Human Pluripotent Stem Cells Enhances HDR-mediated Gene Repair with Customizable Endonucleases. Sci Rep 6: 21264.
- Carlson-Steverner J et al. (2017) Assembly of CRISPR Ribonucleoproteins with Biotinylated Oligonucleotides via an RNA Aptamer for Precise Gene Editing. Nat Commun 8(1): 1711.
- Ma M et al. (2017) Efficient Generation of Mice Carrying Homozygous Double-floxp Alleles Using the Cas9-Avidin/Biotin-donor DNA System. Cell Res 27: 578-581.
- Savic N et al. (2018) Covalent Linkage of the DNA Repair Template to the CRISPR-Cas9 Nuclease Enhances Homology-Directed Repair. Elife 7. Pii:e33761.
- Aird EJ et al. (2018) Increasing Cas9-mediated Homology-Directed Repair Efficiency Through Covalent Tethering of DNA Repair Template. Commun Biol 1: 54.
- Bak R et al. (2018) CRISPR/Cas9 Genome Editing in Human Hematopoietic Stem Cells. Nat Protoc 13(2): 358-376.
- Mitzelfelt KA et al. (2017) Efficient Precision Genome Editing in iPSCs via Genetic Co-targeting with Selection. Stem Cell Rep 8(3): 491-499.
- Ikeda K et al. (2018) Efficient Scarless Genome Editing in Human Pluripotent Stem Cells. Nat Methods 15(12): 1045-1047.
- Martin RM et al. (2019) Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 24(5): 821-828.
- Yiangou L et al. (2019) Method to Synchronize Cell Cycle of Human Pluripotent Stem Cells without Affecting Their Fundamental Characteristics. Stem Cell Rep 12(1): 165-179.
- Haapeniemi E et al. (2018) CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 24: 927-930.
- Ihry RJ et al. (2018) p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat Med 24: 939-946.
- Conti A and Di Micco R (2018) p53 activation: a checkpoint for precision genome editing? Genome Med 10(1): 66.
- Niu D et al. (2017) Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science 357(6357):1303-1307.
- Anderson EM et al. (2015) Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity. J Biotechnol 211: 56-65.
- Kuscu C et al. (2014) Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 32: 677-683.
- Wu X (2014) Target specificity of the CRISPR-Cas9 system. Quant Biol 2(2): 59-70.
- Tsai SQ et al. (2014) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187-197.
- Frock RL et al. (2014) Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33: 179-186.
- Wienert B et al. (2019) Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 364(6437):286-289.
- STEMCELL Technologies (2018) Genome Editing of Human Pluripotent Stem Cells (Document #27084).
- STEMCELL Technologies (2019) Genome Editing of Human Primary T Cells (Document #27155).
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
Related Multimedia
-
Using CRISPR/Cas9 to Model Stem Cell Organization and Dynamics
Publish Date: August 25, 2017 -
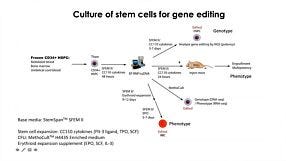
CRISPR-Cas9 Editing of Hematopoietic Stem and Progenitor Cells
Publish Date: June 26, 2018 -
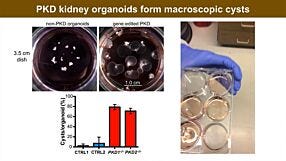
Re-Creating Disease with Kidney Organoids and CRISPR
Publish Date: July 12, 2018

Explore Our Products
-
 Cas9 nuclease for the generation of double-strand breaks in CRISPR-Cas9 genome editing
Cas9 nuclease for the generation of double-strand breaks in CRISPR-Cas9 genome editing -
 cGMP, feeder-free maintenance medium for human ES and iPS cells
cGMP, feeder-free maintenance medium for human ES and iPS cells -
 Custom-designed CRISPR RNA for guide RNA generation in CRISPR-Cas9 genome editing
Custom-designed CRISPR RNA for guide RNA generation in CRISPR-Cas9 genome editing -
 Enhanced green fluorescent protein (eGFP)-tagged Cas9 nuclease for the generation of double-strand breaks in CRISPR-Cas9 genome editing
Enhanced green fluorescent protein (eGFP)-tagged Cas9 nuclease for the generation of double-strand breaks in CRISPR-Cas9 genome editing
