HIV Research Profiles
How Ex Vivo Models Drive Progress in HIV Research
Recently, progress in HIV/AIDS research has been accelerated by the development of new models and assays that use cutting-edge cell isolation technologies to obtain viable primary immune cells directly from fresh blood.
These new ex vivo models enable a closer approximation of the in vivo environment than traditional in vitro approaches. Using these more physiologically relevant models is critical in order to gain a better understanding of the mechanisms of HIV pathogenesis and develop new treatments.1-4
These Research Profiles highlight recent publications that employ a variety of ex vivo models to gain deeper insight into the dynamics of HIV infection. They cover a range of current topics in HIV research, including viral latency, mechanisms for viral resistance in HIV controllers, and new avenues towards potential treatments.
Research Profiles
A Flexible Model of HIV Latency Permitting Evaluation of Many Primary CD4+ T Cell Reservoirs
Lassen KG, Hebbler AM, Bhattacharyya D, Lobritz MA, Green WC. PLoS ONE 7(1): e30176. doi:10.1371/journal.pone.0030176, 2012
Background
The Need for More Physiologically Relevant Models of HIV Latency
HIV-1 establishes latent viral reservoirs in resting memory CD4 + T cells shortly after initial infection, and these persistent reservoirs are a major obstacle to completely eradicating the virus in infected individuals.5,6 Latently infected cells harbor an integrated form of the HIV genome that does not express viral genes, and due to the phenotypically cryptic nature of these cells, their longevity, and their resistance to antiretroviral treatment, there is currently no reliable way to eliminate them from infected subjects.7,8
One potentially effective method of targeting these latent reservoirs is by inducing viral reactivation, allowing the immune system to identify and eliminate latently infected cells. However, research thus far has been constrained by the lack of an ideal model for HIV latency: existing models do not perfectly replicate the state of resting CD4 + T cells in vivo, require lengthy culture periods, or have other limitations.8 In order to achieve greater insight into the precise nature of HIV latency and explore potential treatments, there is now a need for a simple, reproducible and physiologically relevant model.
This research profile highlights a recent PLoS ONE publication by Lassen and colleagues presenting a new model for HIV latency that uses human CD4 + T cells isolated directly from blood.
Experimental Protocol and Results
A Flexible Ex Vivo Model for HIV Latency
In their publication, Lassen et al. describe an improved ex vivo model for studying HIV latency that uses CD4+ T cells isolated from human blood with EasySep™.8 To create this model, Lassen et al. isolate untouched CD4+ T cells using the EasySep™ Human CD4+ T Cell Enrichment Kit, and infect them by spinoculation with novel reporter HIV-1 strains (Figure 1). These infected cells are then used to study HIV latency and gauge the efficacy of various stimuli on inducing latent HIV transcriptional reactivation in primary human CD4+ T cells. Among other applications, the model provides a simple, rapid, and flexible platform to screen candidate antagonists of HIV latency.
One advantage of Lassen et al.'s model is that different CD4+ T cell subsets can be readily substituted at the start of the experiment. In addition to total CD4+ T cells, Lassen et al. isolate memory CD4+ T cells using the EasySep™ Human Memory CD4+ T Cell Kit, and resting memory CD4+ T cells using an EasySep™ custom isolation kit. Carrying out further experiments using these cell subsets, Lassen et al. demonstrate heterogeneity within the memory CD4+ T cell population in response to reactivation agents.8
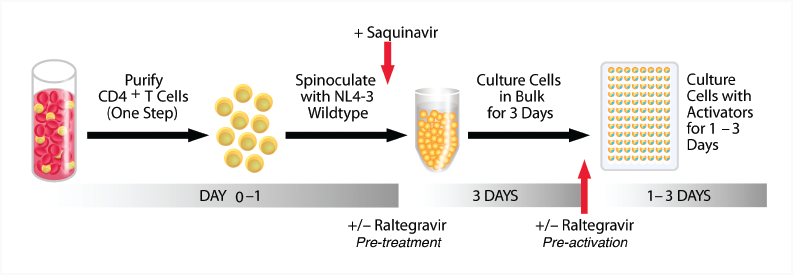
Figure 1. Lassen et al.'s primary cell model of HIV-1 latency. Adapted from Lassen et al. 2012 under the Creative Commons Attribution Licence. Use of this figure does not imply that Lassen et al. or PLoS ONE have endorsed this document.
Conclusion
HIV Latency in the Context of Primary Human Cells
Using highly purified CD4+ T cells isolated by negative selection allows HIV latency to be studied in the context of viable, functional primary cells, thus increasing the physiological relevance of experimental results. The simplicity and flexibility of this model make it amenable to a variety of applications, including high-throughput compound screening, studies of cell subset-specific HIV latency, and research into the molecular mechanisms of latency.8
Ex Vivo T Cell-Based HIV Suppression Assay to Evaluate HIV-Specific CD8+ T Cell Responses
Sáez-Cirión A, Shin SY, Versmisse P, Barré-Sinoussi F, Pancino G. Nat Protoc 5:1033-1041, 2010
Background
Mechanisms of Resistance to Infection in HIV Controllers
HIV Controllers (HIC) are a population of HIV-infected individuals capable of controlling HIV-1 infection to undetectable levels for more than 10 years.9-11 They provide a clue for researchers working on HIV vaccine development, since understanding the basis for their resistance to HIV may make it possible to develop a vaccine capable of recreating the HIC phenotype in typical HIV patients.
Researchers have learned that HIC are characterized by a robust CD8+ T cell response to infection, and in vitro studies have shown that CD8+ T cells in HIC have a number of unusual characteristics, including a high degree of functionality and the capacity to proliferate and generate a multifunctional response to HIV infection.13,14 However, because these studies often used CD8+ T cells activated in vitro with HIV peptides rather than by exposure to infected CD4+ T cells, their relevance in the control of infection is uncertain.14,15
This research profile focuses upon a recent ex vivo assay developed by Asier Sáez-Cirión and colleagues to investigate the underlying mechanisms for HIV resistance by HIC under more physiologically relevant conditions. The assay uses primary human cells isolated by RoboSep™, the fully automated instrument for cell isolation.
Experimental Protocol and Results
The Dynamics of HIV Suppression in HIV Controllers
Much of the world's leading HIV research is conducted at the Institute Pasteur, in the laboratory of Françoise Barré-Sinoussi, President of the International AIDS Society and winner of the 2008 Nobel Prize in Medicine for the discovery of HIV. In order to investigate the basis for HIV suppression in HIC under more physiologically relevant conditions, Asier Sáez-Cirión's research group has developed an ex vivo T cell-based suppression assay that allows HIV-specific CD8+ T cells to be evaluated in terms of their response to infected CD4+ T cells, rather than stimulation by HIV peptides (Figure 2).15
In this study, Sáez-Cirión et al. isolate CD4+ T cells from fresh whole blood or peripheral blood mononuclear cells (PBMCs) by positive selection. CD8+ T cells are isolated from the negative fraction, which is depleted of CD4+ T cells. These CD8+ T cells are isolated by negative selection, (i.e., by targeting unwanted cell types for removal). In order to carry out these steps rapidly, consistently and with minimal human handling, Sáez-Cirión et al. use RoboSep™ for fully automated walk-away cell isolation. Sáez-Cirión's group routinely obtains purities of >95% for CD4+ T cells and CD8+ T cells (see reference 15 for full protocol).
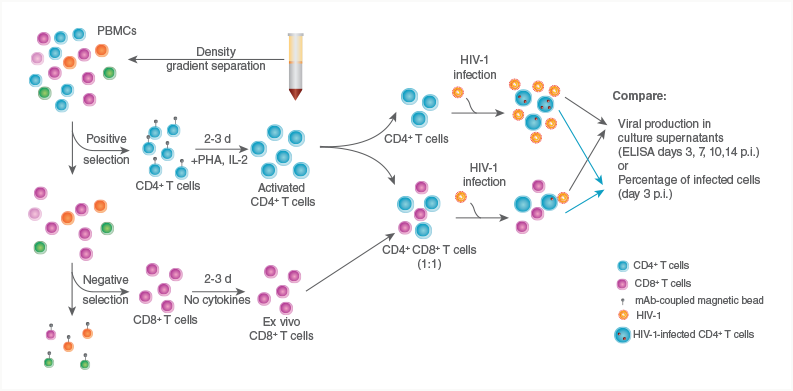
Figure 2. A diagram of the protocol for Sáez-Cirión's T cell-based suppression assay.14 First, CD4+ T cells are separated from PBMCs using the RoboSep™ Human CD4 Positive Selection Kit. CD8+ T cells are then isolated from the remaining fraction using the RoboSep™ CD8 T Cell Enrichment Kit. Isolated CD4+ T cells can then be infected with HIV, and studied both in isolation and in coculture with CD8+ T cells. Adapted by permission from Macmillan Publishers Ltd: Nature Protocols, Sáez-Cirión et al. 2011, copyright 2011.
These purified cells are used in studies investigating HIV infection in pure CD4+ T cell cultures versus infection of CD4+ T cells cocultured with CD8+ T cells. Sáez-Cirión's group provides protocols for assessing levels of infection by either of two proxies: HIV-1 p24 levels in culture supernatants as measured by ELISA, or the percentage of infected CD4+ T cells as assessed by staining for intracellular Gag proteins. This allows the protective effects of CD8+ T cells to be reliably quantified, and the dynamics of infection to be studied in a more physiologically relevant context (i.e., using whole cells rather than peptides). Furthermore, the protocol is readily adaptable to different HIV strains by simply substituting the strain of interest in the viral inoculate.
Do CD8+ T Cells from HIC Suppress HIV Infection?
Sáez-Cirión's research group predicts that ex vivo CD8+ T cells from healthy donor controls should have no significant effect on levels of CD4+ T cell infection, while CD8+ T cells from HIC should be able to reduce viral infection by an order of magnitude or more.15 The results of early experiments support this prediction (Figure 3). The door is now open for HIV labs to further test these hypotheses using the ex vivo T cell-based suppression assay, and to investigate the ultimate mechanistic basis for the protective effects of CD8+ T cells from HIC.
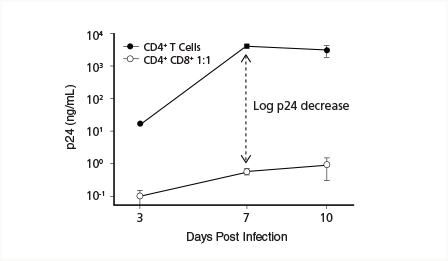
Figure 3. HIV Infection rates in CD4+ T cell monocultures versus CD4+/CD8+ T cell cocultures as measured by HIV-1 p24 levels in culture supernatants. Adapted by permission from Macmillan Publishers Ltd: Nature Protocol, Sáez-Cirión et al. 2011, copyright 2011.
Conclusion
Automated Cell Isolation for Robust, Reproducible HIV Models
This research profile illustrates how automated cell isolation helps create simple, robust and reproducible experimental designs for HIV research. While this leading-edge research is ultimately driven by scientists and their ideas, cell isolation technologies such as RoboSep™ facilitate research and discovery by providing robust automated isolation of multiple primary immune cell populations.
Engineering HIV-Resistant Human CD4+ T Cells with CXCR4-Specific Zinc-Finger Nucleases
Wilen CB. Wang J. Tilton JC. Miller JC. Kim KA. Rebar EJ. Sherrill-Mix SA. Patro SC. Secreto AJ. Jordan AP. Lee G. Kahn J. Aye PP. Bunnell BA. Lackner AA. Hoxie JA. Danet-Desnoyers GA. Bushman FD. Riley JL. Gregory PD. June CH. Holmes MC. Doms RW. PLoS Pathog 7(4): e1002020. doi:10.1371/journal.ppat.1002020, 2011
Background
HIV-1 Requires Specific Surface Markers to Enter Host Cells
HIV-1 gains entry to host T cells in normal individuals by binding first to the CD4 cell surface receptor and then to a co-receptor; either CXCR4 or, more commonly, CCR5.20 Individuals that are homozygous for a CCR5-inactivating mutation (ccr5Δ32) are highly resistant to HIV infection, and infected heterozygotes have a better-than-average prognosis.21-23 CCR5 inhibitors therefore represent one possible approach to HIV treatment, since they may allow normal individuals to reproduce the HIV-resistance phenotype of ccr5Δ32 homozygotes.23
Zinc finger nucleases (ZFNs) provide a powerful way to control the function of specific genes, and ZFNs have been developed to selectively disrupt CCR5 expression in human cells (R5-ZFNs).24,25 ZFNs are artificial restriction enzymes consisting of two main parts: a zinc finger DNA-binding domain, and a DNA cleavage domain. The DNA-binding domains can be engineered to target specific genetic sequences, which are then cut by the DNA cleavage domain. The genetic machinery cells use to repair these cuts is imperfect, and often introduces mutations that inactivate the gene. Disrupting the CCR5 gene in primary human CD4+ T cells using R5-ZFNs is a promising tool for inducing HIV-1 resistance in infected individuals.
Although HIV-1 typically uses CCR5 as a co-receptor to enter cells, some strains are capable of using the cell surface receptor CXCR4 in lieu of CCR5 (X4 HIV), or can utilize either co-receptor (R5X4 HIV).25 There is therefore a need for therapies that target CXCR4 as well as CCR5 to prevent HIV-1 infection.
This research profile focuses on a recent PLoS Pathogens publication in which Wilen et al. present a pair of ZFNs that disrupt CXCR4 in human CD4+ T cells isolated by RosetteSep™, rendering the cells resistant to HIV-1.
Experimental Protocol and Results
Engineering HIV-Resistant Cells by CXCR4 Inactivation
In Wilen et al.'s study, untouched CD4+ T cells from normal donors are isolated from fresh blood using the RosetteSep™ Human CD4+ T Cell Enrichment Cocktail. Wilen et al. then transduce these cells with a viral vector for CXCR4-inactivating ZFNs (X4-ZFNs). In order to test whether this transduction conferred HIV-1 resistance, Wilen et al. challenge the CD4+ T cells in vitro with strains of HIV-1 known to utilize either CCR5 or CXCR4. As expected, transduced CD4+ T cells were resistant to HIV-1 strains known to use CXCR4, but not to those that used CCR5 to enter cells. Furthermore, CD4+ T cells that were homozygous for the CCR5-inactivating ccr5Δ32 allele and transduced with X4-ZFNs were resistant to both types of HIV-1. This suggests that inactivating CCR5 and CXCR4 simultaneously may be an effective treatment for HIV infection.
A Humanized Mouse Model for HIV-Resistant CD4+ T Cells
To investigate whether X4-ZFNs conferred HIV-1 resistance in vivo as well as in vitro, Wilen et al. injected a group of mice with X4-ZFN-treated human CD4+ T cells isolated using RosetteSep™, and a separate group of mice with R5-ZFN-treated human CD4+ T cells. 28 days later, they injected all mice with autologous CD4+ T cells previously infected with X4 HIV (a strain of HIV that uses CXCR4 to enter cells).
After 34 days, Wilen et al. isolated CD4+ T cells from mouse peripheral blood and spleens by positive selection using RoboSep™. They then compared the frequency of CXCR4 disruption in HIV-infected mice compared to control mice. CXCR4 disruption was significantly greater in HIV-infected mice, which suggests that HIV infection caused selection in favor of X4-ZFN-transduced (i.e. X4 HIV-resistant) cells (Figure 4).
By monitoring human CD4+ T cell counts in both groups of mice, Wilen et al. found that treatment with X4-ZFN conferred significant protection to HIV, but that this protection waned over time. Wilen et al. sequenced viral RNA isolated from mouse plasma after the experiment, and identified a mutation in viral Env isolated from X4-ZFN mice, which likely enabled HIV to use CCR5. This suggests that the virus rapidly evolved in vivo to use CC5R in place of CXCR4, which explains the transient nature of HIV resistance in their model.
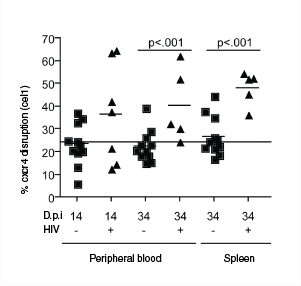
Figure 4. Selection for X4-ZFN-transduced CD4+ T cells in X4 HIV-infected mice (+), but not control mice (-). Adapted from Wilen et al. 2011 under the Creative Commons Attribution Licence. Use of this figure does not imply that Wilen et al. or PLoS Pathogens have endorsed this document.
Conclusion
Progress towards Conferring Permanent HIV Resistance
Gene modification using ZFNs is a promising avenue towards engineering permanent HIV resistance. Ex vivo cell-based assays and humanized mouse studies are an important way to evaluate the efficacy of this type of treatment prior to human trials, and using primary human cells isolated directly from blood maximizes the physiological relevance of these approaches. RosetteSep™ provides a means of isolating untouched immune cells from whole blood in a single step, and RoboSep™ allows fully automated manipulation of infectious samples, minimizing human exposure and reducing hands-on time for technicians.
The Antiviral Factor APOBEC3G Enhances the Recognition of HIV-Infected Primary T Cells by NK Cells
Norman JM, Mashiba M, McNamara LA, Onafuwa-Nuga A, Chiari-Fort E, Shen W, Collens KL. Nat Immunol 12(10): 975-986, 2011
Background
Molecular Interactions Between HIV-1 and Host Cells
HIV infection entails complex molecular interactions between host cells and the virus, and some such interactions involve an enzyme called APOBEC3G (A3G). A3G inhibits HIV replication by disrupting cDNA synthesis,17 in part through deaminating cytosine residues on single-stranded viral DNA, changing them to uracil. This leads to hypermutation during cDNA synthesis, resulting in viral inactivation.18 HIV-1 counteracts this by targeting A3G for proteosomal degradation by viral infectivity factor (Vif). The molecular conflict between host and virus can increase the rate of of sublethal viral mutations, which accelerates the evolution of HIV-1 and helps it evade the immune response and antiretroviral drugs.19
Evidence suggests that cytidine deaminases such as A3G also help trigger the innate immune response to infection, but the underlying mechanisms for this effect are still unclear. In their recent Nature Immunology paper, Norman et al. use an ex vivo HIV infection model based on PBMCs and natural killer (NK) cells isolated from blood using EasySep™. The authors elucidate the mechanism by which both host and viral proteins interact to initiate the DNA damage response upon HIV infection. This interaction initiates a suicide response whereby increased expression of NK-cell-activating ligands sensitizes the HIV-infected cell to NK-cell-mediated killing.
Experimental Protocol and Results
A3G and Vif Modulate NKG2D Ligand Expression in HIV-Infected Cells
A number of cell surface molecules determine the interactions between NK cells and HIV-infected T cells. The expression of these molecules is modulated by the host protein A3G and UNG2, as well as the viral proteins Nef, Vif, and Vpr. To elucidate the effects of these proteins, Norman et al. use a model in which each protein, along with other variables, can be independently manipulated in the context of ex vivo primary cells.
Norman et al. deplete PBMCs of CD56+ cells using EasySep™ and infect them with an HIV-1 strain carrying a Vif-inactivating mutation. In so doing, they demonstrate that inactivating Vif increases NKG2D ligand (NKG2D-L) expression by host cells in the presence of HIV-1. Since Vif inactivates A3G, Norman et al. next investigate whether A3G is responsible for the upregulation of NKG2D-L on infected host T cells. They show that variation in the quantity of A3G correlates with NKG2D-L expression, and through further experiments show that A3G upregulates NKG2D-L by activating the ATM (DNA damage response) pathway through interactions of UNG2 and Vpr.
A3G Enhances the Recognition of HIV-Infected Primary Cells by NK Cells
To determine the effect of NKG2D-L upregulation on NK-cell-mediated lysis of HIV-1 infected cells, Normal et al. isolate CD56+ NK cells from PBMCs using EasySep™. They culture the autologous NK cells with HIV-infected T cells, and establish that target cell expression levels of NKG2D-L dictates the sensitivity for NK-mediated lysis. Next, Norman et al. assess the effects of Vif inactivation and shRNA-mediated A3G knockdown on T cell survival, and show that inactivating Vif results in significantly decreased survival of infected T cells, while shRNA knockdown of A3G significantly increases their survival, even in the absence of Vif. These results suggest that A3G sensitizes infected cells to lysis by NK cells by upregulating NKG2D-L, and that Vif counteracts this effect by degrading A3G.
Conclusion
Investigating the Underlying Mechanisms for Host-Virus Interactions
Investigating molecular mechanisms for host-virus interactions can be facilitated through elegant experimental models allowing both immune cells and viral strains to be isolated and manipulated independently. The physiological relevance of these models is maximized through the use of ex vivo primary cells with intact functions isolated directly from blood. EasySep™ cell isolation kits allow the isolation of primary human cells for immediate use in microcosm studies of host-virus interactions.
References
- Thomas C. Nat Med 15: 855-859, 2009
- Moir S, et al. Ann Rev Pathol 6: 223-248, 2011
- Virgin HW, Walker BD. Nature 464: 224-230, 2010
- Dieffenbach CW, et al. Ann Intern Med 154: 1-7, 2011
- Chun TW, et al. Nature 387: 183-188, 1997
- Wong JK, et al. Science 278: 1291-1295, 1997
- Chomont N, et al. Nat Med 15(8): 893-900, 2009
- Lassen KG, et al. PloS One 7(1): e30176.doi:10.1371/journal.pone.0030176, 2012
- Sáez-Cirión A, et al. Trends Immunol 28: 532-540, 2007
- Deeks SG, Walker BD. Immunity 27: 406-416, 2007
- O'Connell KA, et al. Trends Pharmacol Sci 30: 631-637, 2009
- Sáez-Cirión A, et al. Nat Protoc 5: 1033-1041, 2010
- Betts MR, et al. Blood 107: 4871-4879, 2006
- Migueles SA, et al. Nat Immunol. 3:1061-1068 2002
- Sáez-Cirión A, et al. Nat Protoc. 5:1033-1041 2010
- Sáez-Cirión A, et al. Blood. 118:955-964 2011
- Peng G, et al. J Exp Med 203: 41-46, 2006
- Norman JM, et al. Nat Immun 12: 975-983, 2011
- Jern P, et al. PLoS Pathog 5(4): e1000367, 2009
- Tilton JC, Doms RW. Antiviral Res 85: 91-100, 2010
- Samson M, et al. Nature 382: 722-5, 1996
- Dean M, et al. Science 273: 1856-1862, 1996
- Huang Y, et al. Nat Med 2: 1240-3, 1996
- Perez EE, et al. Nat Biotechnol 26: 808-16, 2008
- Wilen CB, et al. PLoS Pathog 7(4): e1002020. doi:10.1371/journal.ppat.1002020, 2011
