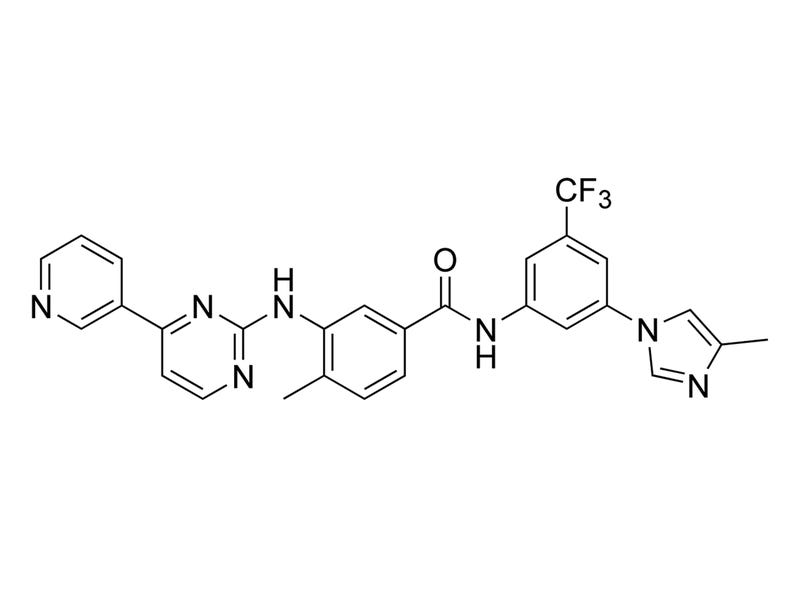
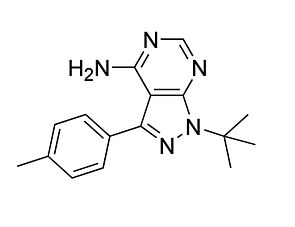
Nilotinib
Tyrosine kinase inhibitor; Inhibits BCR/ABL and ABL
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (5)
Activity-based kinase profiling of approved tyrosine kinase inhibitors.
Genes to cells : devoted to molecular & cellular mechanisms 2013
Abstract
The specificities of nine approved tyrosine kinase inhibitors (imatinib, dasatinib, nilotinib, gefitinib, erlotinib, lapatinib, sorafenib, sunitinib, and pazopanib) were determined by activity-based kinase profiling using a large panel of human recombinant active kinases. This panel consisted of 79 tyrosine kinases, 199 serine/threonine kinases, three lipid kinases, and 29 disease-relevant mutant kinases. Many potential targets of each inhibitor were identified by kinase profiling at the K(m) for ATP. In addition, profiling at a physiological ATP concentration (1 mm) was carried out, and the IC(50) values of the inhibitors against each kinase were compared with the estimated plasma-free concentration (calculated from published pharmacokinetic parameters of plasma C(trough) and C(max) values). This analysis revealed that the approved kinase inhibitors were well optimized for their target kinases. This profiling also implicates activity at particular off-target kinases in drug side effects. Thus, large-scale kinase profiling at both K(m) and physiological ATP concentrations could be useful in characterizing the targets and off-targets of kinase inhibitors.
Extended kinase profile and properties of the protein kinase inhibitor nilotinib.
Biochimica et biophysica acta 2010
Abstract
As a drug used to treat imatinib-resistant and -intolerant, chronic and advanced phase chronic myelogenous leukaemia, nilotinib is well characterised as a potent inhibitor of the Abl tyrosine kinase activity of wild-type and imatinib-resistant mutant forms of BCR-Abl. Here we review the profile of nilotinib as a protein kinase inhibitor. Although an ATP-competitive inhibitor of Abl, nilotinib binds to a catalytically inactive conformation (DFG-out) of the activation loop. As a consequence of this, nilotinib exhibits time-dependent inhibition of Abl kinase in enzymatic assays, which can be extrapolated to other targets to explain differences between biochemical activity and cellular assays. Although these differences confound assessment of kinase selectivity, as assessed using a combination of protein binding and transphosphorylation assays, together with cellular autophosporylation and proliferation assays, well established kinase targets of nilotinib in rank order of inhibitory potency are DDR-1textgreaterDDR-2textgreaterBCR-Abl (Abl)textgreaterPDGFRalpha/betatextgreaterKITtextgreaterCSF-1R. In addition nilotinib has now been found to bind to both MAPK11 (p38beta) and MAPK12 (p38alpha), as well as with very high affinity to ZAK kinase. Although neither enzymatic nor cellular data are yet available to substantiate the drug as an inhibitor of ZAK phosphorylation, modeling predicts that it binds in an ATP-competitive fashion.
Transcriptional profiling of the dose response: a more powerful approach for characterizing drug activities.
PLoS computational biology 2009 SEP
Abstract
The dose response curve is the gold standard for measuring the effect of a drug treatment, but is rarely used in genomic scale transcriptional profiling due to perceived obstacles of cost and analysis. One barrier to examining transcriptional dose responses is that existing methods for microarray data analysis can identify patterns, but provide no quantitative pharmacological information. We developed analytical methods that identify transcripts responsive to dose, calculate classical pharmacological parameters such as the EC50, and enable an in-depth analysis of coordinated dose-dependent treatment effects. The approach was applied to a transcriptional profiling study that evaluated four kinase inhibitors (imatinib, nilotinib, dasatinib and PD0325901) across a six-logarithm dose range, using 12 arrays per compound. The transcript responses proved a powerful means to characterize and compare the compounds: the distribution of EC50 values for the transcriptome was linked to specific targets, dose-dependent effects on cellular processes were identified using automated pathway analysis, and a connection was seen between EC50s in standard cellular assays and transcriptional EC50s. Our approach greatly enriches the information that can be obtained from standard transcriptional profiling technology. Moreover, these methods are automated, robust to non-optimized assays, and could be applied to other sources of quantitative data.
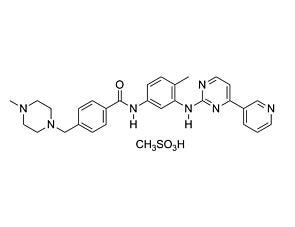
Related Products
Related Products
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
