Make more informed purchasing decisions with our new product availability and delivery estimate feature, now available on all product pages, in your cart, and during checkout.
Sign In
New to STEMCELL?
Register for an account to quickly and easily purchase products online and for one-click access to all educational content.
Thank you for your interest in this product.
Please provide us with your contact information and your local representative
will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
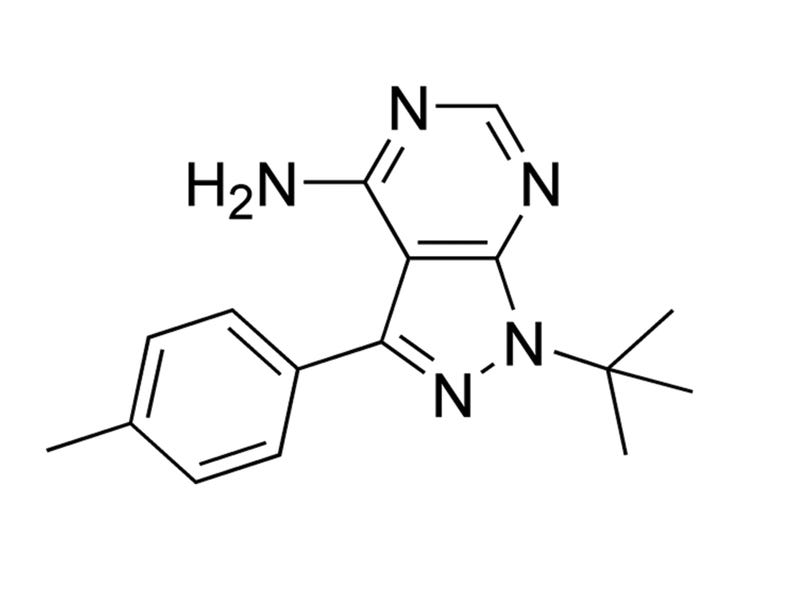
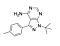
PP1 is a reversible inhibitor of the SRC family of tyrosine kinases. It inhibits LCK, FYN, HCK and SRC with IC₅₀ values of 5, 6, 20 and 170 nM, respectively (Hanke et al.). It is relatively selective for SRC family kinases versus other kinases, inhibiting epidermal growth factor receptor (EGFR), janus-activated kinase 2 (JAK2) and zeta-chain-associated protein kinase 70 (ZAP70) with IC₅₀ values of 0.25, > 50, and > 100 μM, respectively, and KIT, platelet-derived growth factor receptor (PDGFR), and RET tyrosine kinase in the 75 - 100 nM range (Carlomagno et al. ; Tatton et al.; Waltenberger et al.; Hanke et al.). PP1 also blocks TGF-β-mediated cellular responses by directly inhibiting type I TGF-β receptors (IC₅₀ = 50 nM; Ungefroren et al.; Maeda et al.).
REPROGRAMMING
· Enables reprogramming of mouse embryonic fibroblasts to induced pluripotent stem cells in the absence of reprogramming factor SOX2 (Staerk et al.; Ma et al.).
CANCER RESEARCH
· Blocks TGF-β-mediated migration of primary non-small cell lung carcinoma cells and pancreatic ductal adenocarcinoma cell lines (Bartscht et al.).
· Induces apoptosis in non-small cell lung cancer cell lines (Zhang et al.).
Cell Type
Cancer Cells and Cell Lines, Pluripotent Stem Cells
This product is designed for use in the following research area(s) as part
of the highlighted workflow stage(s). Explore these workflows to learn more about the other products we
offer to support each research area.
Progress in the reprogramming of somatic cells.
Ma T et al.
Circulation research 2013
Abstract
Pluripotent stem cells can differentiate into nearly all types of cells in the body. This unique potential provides significant promise for cell-based therapies to restore tissues or organs destroyed by injuries, degenerative diseases, aging, or cancer. The discovery of induced pluripotent stem cell (iPSC) technology offers a possible strategy to generate patient-specific pluripotent stem cells. However, because of concerns about the specificity, efficiency, kinetics, and safety of iPSC reprogramming, improvements or fundamental changes in this process are required before their effective clinical use. A chemical approach is regarded as a promising strategy to improve and change the iPSC process. Dozens of small molecules have been identified that can functionally replace reprogramming factors and significantly improve iPSC reprogramming. In addition to the prospect of deriving patient-specific tissues and organs from iPSCs, another attractive strategy for regenerative medicine is transdifferentiation-the direct conversion of one somatic cell type to another. Recent studies revealed a new paradigm of transdifferentiation: using transcription factors used in iPSC generation to induce transdifferentiation or called iPSC transcription factor-based transdifferentiation. This type of transdifferentiation not only reveals and uses the developmentally plastic intermediates generated during iPSC reprogramming but also produces a wide range of cells, including expandable tissue-specific precursor cells. Here, we review recent progress of small molecule approaches in the generation of iPSCs. In addition, we summarize the new concept of iPSC transcription factor-based transdifferentiation and discuss its application in generating various lineage-specific cells, especially cardiovascular cells.
The Src family kinase inhibitors PP2 and PP1 effectively block TGF-beta1-induced cell migration and invasion in both established and primary carcinoma cells.
Bartscht T et al.
Cancer chemotherapy and pharmacology 2012
Abstract
PURPOSE: We have previously demonstrated that in pancreatic ductal adenocarcinoma (PDAC)-derived cell lines, the common Src family kinase inhibitors PP2 and PP1 effectively inhibited morphologic alterations associated with TGFβ1-mediated epithelial-to-mesenchymal transition (EMT) by blocking the kinase activity of the TGF-β type I receptor ALK5 rather than Src (Ungefroren et al. in Curr Cancer Drug Targets 11:524, 2011). In this report, the ability of PP2 and PP1, the more specific Src inhibitor SU6656, and the ALK5 inhibitor SB431542 to functionally block TGF-β1-dependent EMT and cell motility in established PDAC (Panc-1, Colo 357) and primary NSCLC (Tu459) cell lines were investigated. METHODS: The effects of PP2, PP1, SU6656, and SB431542 on TGF-β1-dependent cell scattering/EMT, cell migration/invasion, and expression of invasion-associated genes were measured by using the real-time cell analysis assay on the xCELLigence system and quantitative real-time RT-PCR, respectively. RESULTS: In all three cell lines tested, PP1, PP2, and SB431542 effectively blocked TGF-β1-induced cell scattering/EMT, migration, and invasion and in Colo 357 cells inhibited the induction of the invasion-associated MMP2 and MMP9 genes. In contrast, SU6656 only blocked TGF-β1-induced invasion in Panc-1 and Tu459 but not Colo 357 cells. PP1, and to a greater extent PP2, also inhibited the high spontaneous migratory activity of Panc-1 cells expressing a kinase-active ALK5 mutant. CONCLUSIONS: These data provide evidence that PP2 and PP1 are powerful inhibitors of TGF-β-induced cell migration and invasion in vitro and directly target ALK5. Both agents may be useful as dual TGF-β/Src inhibitors in experimental therapeutics to prevent metastatic spread in late-stage PDAC and NSCLC.
Pan-Src family kinase inhibitors replace Sox2 during the direct reprogramming of somatic cells.
Staerk J et al.
Angewandte Chemie (International ed. in English) 2011
Tyrosine kinase inhibitor; Inhibits ABL, SRC, LCK and YES
Item added to your cart
PP1
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.