Evaluation of Genome Editing
- Document # 27126
- Version 1.0.2
- Feb 2022
Overview of Screening and Verification Strategies
Introduction
CRISPR-Cas9 is a system for creating specific and accurate edits to
the genome of a wide variety of target cell types. Depending on
experimental design, genome editing with CRISPR-Cas9 has many
applications, such as creating specific point mutations or mutation
corrections, “knock-in” gene insertions, whole gene deletions, or
short disruptive insertions or deletions (INDELs).1 It is important to
assess genome-edited cells to verify that the alteration of interest
occurred without inadvertently affecting off-target loci. This
document provides an overview of commonly used methods to
detect, verify, and quantify CRISPR-Cas9-mediated genome editing.
Generally, a screening assay is first performed to detect the
presence of a genetic alteration. This can be accomplished
using a heterogeneous population of cells 48 - 72 hours after
CRISPR-Cas9 ribonucleoprotein (RNP) delivery and/or clonal
(isogenic) subcultures. Screening alone may be sufficient to
select an effective guide RNA (or crRNA) in the first instance
and, depending on the assay chosen, can be informative of the
relative proportion of possible editing outcomes observed,
i.e. heterozygous mutant (INDEL or single base edit),
homozygous mutant, compound heterozygous mutant, or
wild-type. However, when the edited cells are to be used in
downstream assays, screening should be followed by a more
detailed sequence assessment in positive populations or
clones, both at the target locus and at potential off-target loci,
to ultimately obtain a pure population of cells containing only
the alteration of interest.
This bulletin describes four strategies for screening edited cells
based on mismatch cleavage, Sanger sequencing analysis,
polymerase chain reaction (PCR) amplicon length, and restriction
endonuclease pattern. While the mismatch cleavage assay and
Sanger sequencing analysis can be used to screen for edits from
most experimental designs, PCR or restriction endonuclease-based
assays require that screening tools are incorporated into the project
design. We also briefly describe ways to confirm on- and off-target
effects at the sequence level.
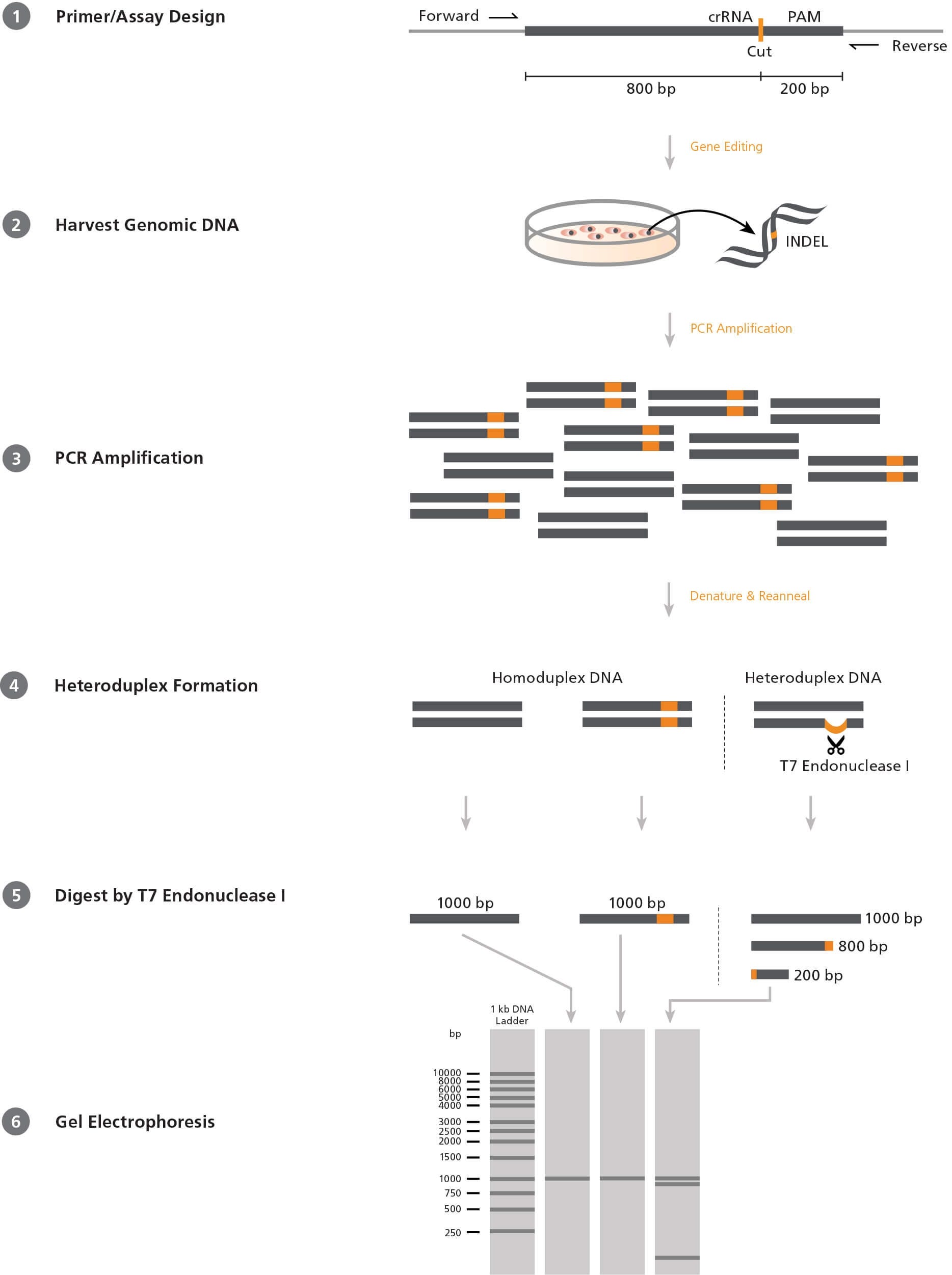
Figure 1. Mismatch Cleavage Assay Using T7 Endonuclease I
1) Forward and reverse PCR primers are designed to flank the target CRISPR-Cas9 site in an offset manner, e.g. 200 and 800 base pairs (bp) on either side. 2) After CRISPR-Cas9 editing, genomic DNA is extracted from the cells. 3) The target region is amplified by PCR using the above primers. 4) PCR products are denatured and reannealed; DNA from edited cells may reanneal with DNA from non-edited (wild-type) cells to create a heteroduplex. 5) ArciTect™ T7 Endonuclease I will cleave single-strand DNA at heteroduplex structures > 2 bp. 6) Due to the offset nature of the primers, the resulting fragments will be of different lengths and can be resolved on an agarose gel. The relative amount of cut fragments detectable on the gel thereby gives an estimate of the mutation frequency within the cell population.
Screening Assays
The most commonly used system to screen for INDELs in a
heterogeneous cell population is the mismatch cleavage assay.
This assay utilizes a DNA endonuclease with single-strand cleavage
activity specific to heteroduplex structures (Figure 1). To perform
the mismatch cleavage assay, the target region is first amplified
from genomic DNA by PCR using primers that are offset to the
desired Cas9 cut site (e.g. targeting sequence 200 and 800 base
pairs [bp] up- and downstream respectively). The PCR-generated
amplicons are then denatured and reannealed randomly. Where
present, INDEL-containing DNA will reanneal with wild-type DNA
into heteroduplex (“mismatched”) structures, generating a singlestrand
loop of non-complementarity. An endonuclease such as
ArciTect™ T7 Endonuclease I is then added, which cleaves at the
loop to generate two shorter DNA fragments. The fragments are
resolved on an agarose gel and the relative proportion of uncut
and cut fragments provides a semi-qualitative estimate of the
genome editing frequency within the population.
T7 Endonuclease I is commonly used in the mismatch cleavage
assay. It is a single-strand-specific endonuclease derived from
bacteriophage, with high sensitivity to mismatches of at least
2 base pairs. Since it requires heteroduplex DNA to be formed,
homozygous mutations will not be detected in clonal cell lines
unless the PCR reaction is spiked with wild-type DNA. Due to
these caveats, the editing efficiency determined by this method
is generally an underestimate. Overall, the mismatch cleavage
assay is a useful tool to quickly screen for the presence of INDELs
and provides relative editing frequency between experimental
conditions.
Sanger sequencing can provide a relatively quantitative estimate of
editing efficiency in pooled cell populations. Primers flanking the
target locus are used to amplify the region and those PCR products
are submitted for sequencing. It is important to include a wildtype
control, such as the parental cell line, for comparison. Once
sequencing data is obtained, the readout can be visually inspected
and/or editing efficiencies can be calculated. Recently, a number of open-source software programs were developed to analyze Sanger sequencing traces generated from edited cell populations,
such as the Tracking of INDELs by Decomposition (TIDE), Tracking
of Insertion, DEletions and Recombination events (TIDER), INDEL
Detection Amplicon Analysis (IDAA), or Inference of CRISPR Editing
(ICE).2-5 These tools use slightly different algorithms to compare
PCR-amplified and Sanger-sequenced DNA from both edited and
unedited (control) populations and provide a detailed analysis of
CRISPR editing outcomes.
PCR strategies can sometimes be used to detect positive editing
events, particularly for large insertions or deletions. For example, if
the experimental goal is to delete a large coding region, then PCR
using primers located outside of the target region will generate
a smaller amplicon in cells where the deletion has occurred.
Similarly, if homology-directed repair (HDR) is used to “knock-in”
a DNA sequence, then PCR will generate a larger amplicon in cells
where the insertion has occurred. The above strategy is limited to
experimental designs that generate large changes (> 50 bp) in DNA
length at the target locus.
When genome editing also incorporates a change at a restriction
endonuclease cut site(s), editing efficiency can be estimated by
assessing the change in the restriction endonuclease digestion
pattern. To do this, guide RNAs are designed to overlap
a restriction site; when INDELs occur, the restriction site is
destroyed. Genome editing can then be assessed by the absence
of restriction endonuclease digestion in PCR-amplified copies
of the target locus. Similarly, a restriction site may be created
(or destroyed) due to a targeted substitution created via HDR.
The difficulty with this method is finding a restriction site that
perfectly overlaps with a favorable mutation target site. A recent
refinement to this approach involves the use of primers that contain
mismatches, resulting in creation of a restriction site in one of the
amplified templates.6
Verification of Genome Editing in Clonal Cell Lines
Regardless of the preliminary tactic used to screen for editing
events, researchers should sequence deeply across the target locus
to confirm the desired mutation is present at the desired allele
frequency in potential positive clones.
Sanger sequencing represents the most accessible readout of the
coding alteration and is a widely available service. As outlined
above, primers flanking the target locus are used to amplify the
region of interest and those PCR products are submitted for
sequencing. In addition, sequencing can be used to ensure that
clones with any potential off-target edits can be eliminated from
further characterization. Primers designed around predicted
off-target sites (i.e. those identified by guide RNA design
programs)1 can be used to amplify samples for sequencing.
Determining the nature of the changes introduced throughout the
genome after genomic editing is a necessary step to ensure validity
of the experiment prior to functional assays.
Although it is cost-prohibitive, next-generation sequencing (NGS)
can provide sequence data of customizable target regions or of the
entire genome (whole genome sequencing; WGS). Targeted
deep-sequencing can be used to accurately assess editing
efficiencies in cell pools and/or to simultaneously validate
clonogenicity and the absence of off-target mutations with high
confidence.7,8 Unlike Sanger sequencing or targeted NGS, WGS
does not require specific sequencing primers and pre-determined
loci. A library prepared from the genomic sample is amplified,
sequenced, and mapped to a reference genome for variant
detection. Algorithms such as CRISPResso (http://crispresso.rocks)9
or CRISPR Genome Analyzer (CRISPR-GA; http://crispr-ga.net)10
can be used to analyze NGS data. The NGS method provides DNA
sequences at the target locus, homologous loci, and all possible
sites throughout the genome in a non-biased manner.
The above assays describe ways to evaluate editing at the
genomic level. Once positive clones are identified, functional
studies comparing multiple edited and unedited clones can
identify mechanistic roles for the gene products. Through
rigorous assessment of on- and off-target sites, the researcher can
confidently attribute phenotypic changes to the induced alteration.
Related Product Information
References
- STEMCELL Technologies. (2018) Technical Bulletin: Design Considerations for the ArciTect™ CRISPR-Cas9 Genome Editing System (Document #27083).
- Brinkman EK et al. (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42(22): e168.
- Brinkman EK et al. (2018) Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res 46(10): e58.
- Yang Z et al. (2015) Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res 43(9): e59.
- Hsiau T et al. (2018) Inference of CRISPR edits from Sanger trace data. bioRxiv. DOI: 10.1101/251082.
- Hodgens C et al. (2017) indCAPS: A tool for designing screening primers for CRISPR/Cas9 mutagenesis events. PLoS One 12(11): e0188406.
- Bell C et al. (2014) A high-throughput screening strategy for detecting CRISPR-Cas9 induced mutations using next-generation sequencing. BMC Genomics 15: 1002.
- Sentmanat MF et al. (2018) A survey of validation strategies for CRISPR-Cas9 editing. Sci Rep 8(1): 888.
- Pinello L et al. (2016) Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol 34(7): 695–7.
- Güell M et al. (2014) Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics 30(20): 2968–70.
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration

