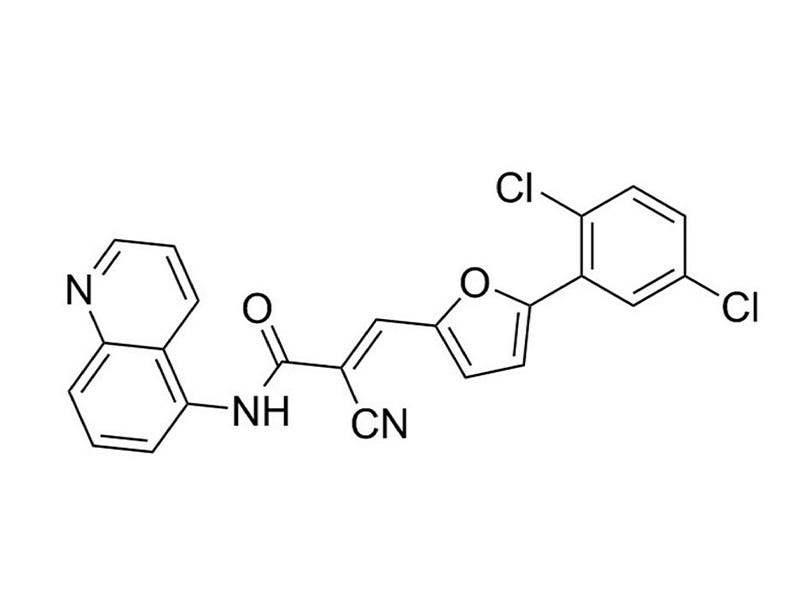
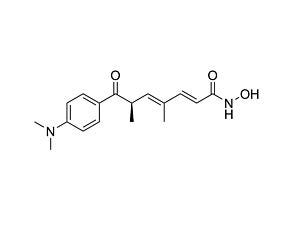
AGK2
Epigenetic modifier; Inhibits SIRT2 histone deacetylase
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Publications (4)
The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease.
Nature medicine 2015 MAR
Abstract
The ketone bodies β-hydroxybutyrate (BHB) and acetoacetate (AcAc) support mammalian survival during states of energy deficit by serving as alternative sources of ATP. BHB levels are elevated by starvation, caloric restriction, high-intensity exercise, or the low-carbohydrate ketogenic diet. Prolonged fasting reduces inflammation; however, the impact that ketones and other alternative metabolic fuels produced during energy deficits have on the innate immune response is unknown. We report that BHB, but neither AcAc nor the structurally related short-chain fatty acids butyrate and acetate, suppresses activation of the NLRP3 inflammasome in response to urate crystals, ATP and lipotoxic fatty acids. BHB did not inhibit caspase-1 activation in response to pathogens that activate the NLR family, CARD domain containing 4 (NLRC4) or absent in melanoma 2 (AIM2) inflammasome and did not affect non-canonical caspase-11, inflammasome activation. Mechanistically, BHB inhibits the NLRP3 inflammasome by preventing K(+) efflux and reducing ASC oligomerization and speck formation. The inhibitory effects of BHB on NLRP3 are not dependent on chirality or starvation-regulated mechanisms like AMP-activated protein kinase (AMPK), reactive oxygen species (ROS), autophagy or glycolytic inhibition. BHB blocks the NLRP3 inflammasome without undergoing oxidation in the TCA cycle, and independently of uncoupling protein-2 (UCP2), sirtuin-2 (SIRT2), the G protein-coupled receptor GPR109A or hydrocaboxylic acid receptor 2 (HCAR2). BHB reduces NLRP3 inflammasome-mediated interleukin (IL)-1β and IL-18 production in human monocytes. In vivo, BHB or a ketogenic diet attenuates caspase-1 activation and IL-1β secretion in mouse models of NLRP3-mediated diseases such as Muckle-Wells syndrome, familial cold autoinflammatory syndrome and urate crystal-induced peritonitis. Our findings suggest that the anti-inflammatory effects of caloric restriction or ketogenic diets may be linked to BHB-mediated inhibition of the NLRP3 inflammasome.
NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells.
The Journal of clinical investigation 2014 DEC
Abstract
High aldehyde dehydrogenase (ALDH) activity is a marker commonly used to isolate stem cells, particularly breast cancer stem cells (CSCs). Here, we determined that ALDH1A1 activity is inhibited by acetylation of lysine 353 (K353) and that acetyltransferase P300/CBP-associated factor (PCAF) and deacetylase sirtuin 2 (SIRT2) are responsible for regulating the acetylation state of ALDH1A1 K353. Evaluation of breast carcinoma tissues from patients revealed that cells with high ALDH1 activity have low ALDH1A1 acetylation and are capable of self-renewal. Acetylation of ALDH1A1 inhibited both the stem cell population and self-renewal properties in breast cancer. Moreover, NOTCH signaling activated ALDH1A1 through the induction of SIRT2, leading to ALDH1A1 deacetylation and enzymatic activation to promote breast CSCs. In breast cancer xenograft models, replacement of endogenous ALDH1A1 with an acetylation mimetic mutant inhibited tumorigenesis and tumor growth. Together, the results from our study reveal a function and mechanism of ALDH1A1 acetylation in regulating breast CSCs.
Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity.
Nature cell biology 2010 JUL
Abstract
Autophagy is characterized by the sequestration of bulk cytoplasm, including damaged proteins and organelles, and delivery of the cargo to lysosomes for degradation. Although the autophagic pathway is also linked to tumour suppression activity, the mechanism is not yet clear. Here we report that cytosolic FoxO1, a forkhead O family protein, is a mediator of autophagy. Endogenous FoxO1 was required for autophagy in human cancer cell lines in response to oxidative stress or serum starvation, but this process was independent of the transcriptional activity of FoxO1. In response to stress, FoxO1 was acetylated by dissociation from sirtuin-2 (SIRT2), a NAD(+)-dependent histone deacetylase, and the acetylated FoxO1 bound to Atg7, an E1-like protein, to influence the autophagic process leading to cell death. This FoxO1-modulated cell death is associated with tumour suppressor activity in human colon tumours and a xenograft mouse model. Our finding links the anti-neoplastic activity of FoxO1 and the process of autophagy.
Related Products
Related Products
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
