Intestinal Organoid Culture
The luminal surface of the mammalian intestinal tract consists of a single layer of rapidly self-renewing epithelial cells that can be completely replenished within five days
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration

- Document # MRDX20358
- Version 1.0.0
- Apr 2015
The Intestinal Epithelium
The luminal surface of the mammalian intestinal tract consists of a single layer of rapidly self-renewing epithelial cells that can be completely replenished within five days.1 The epithelium is divided into two distinct regions that are designated the crypt and villus domains. The crypt domain is located at the most basal part of the intestinal epithelium and is in direct contact with the basement membrane. The crypts contain the intestinal stem cells (ISCs). Conversely, the villus domain is located at the apical surface of the intestinal epithelium and is composed of several differentiated cell types.1
There are five predominant cell types that make up the intestinal epithelium.1 The proliferative stem cells are located at the terminal region of the crypt and are flanked by Paneth cells, which secrete cytokines that help maintain the stem cell niche. The Paneth cells are also responsible for the secretion of anti-microbial peptides.1 The stem cells divide symmetrically and either renew the stem cell population at the crypt base or begin differentiating by becoming part of the rapidly dividing transit amplifying population of the crypt, depending on the specific location of the dividing stem cell within the crypt. The transit amplifying cells move apically towards the villi where they will terminally differentiate into absorptive enterocytes, hormone-secreting enteroendocrine cells or mucus-secreting goblet cells. The mature epithelial cells move up the villi where they will eventually exfoliate from the villus tip into the lumen.1
It is only recently that the proliferative stem cell population of the adult intestine has been definitively identified through lineage-tracing experiments as being composed of the columnar cells that are intercalated with the Paneth cells at the crypt base.2 These cells express LGR5, a protein in the WNT signaling pathway, and can give rise to all cell types of the adult intestine. Further studies have demonstrated that populations of stem cells located outside of the crypt base, specifically cells in the +4 position of the crypt, can revert into multipotent stem cells upon intestinal damage.3 These findings demonstrate the plasticity of the stem and progenitor cells in the intestinal tissue that is necessary to maintain homeostasis in the harsh environment of the intestinal lumen.
The LGR5+ stem cells retain their capacity to self-renew and regenerate the intestinal epithelium primarily due to their position within a stem cell niche.4 The intestinal niche has been well characterized and shown to consist of spatial gradients of high WNT and Epithelial Growth Factor (EGF), while Bone Morphogenetic Protein (BMP) signals are inhibited.5 Study of this defined niche has led to a model of neutral drift. Within this model, stem cells divide symmetrically within a spatially restricted niche. This proliferation necessarily results in the removal of some stem cells from the niche while the remaining stem cells retain their position at the crypt base and consequently maintain their “stemness”.6 Over time, the stem cells in a given crypt drift toward clonality as the progeny of neighboring stem cells are excluded from the niche.
The detailed knowledge that has previously been collected regarding the cell lineages and functions of the intestinal epithelium coupled with the continually renewing nature of this tissue make it an attractive model system to study the general characteristics of epithelial tissues and adult stem cells, as well as the specific characteristics of the intestinal lining relevant to disease and drug design.
The History of Intestinal Organoids
Studies using genetic manipulations of mouse models and immortalized cell lines, such as the CACO-2 adenocarcinoma line, have yielded significant understanding of intestinal function, including the role of the individual cell types that make up this epithelial tissue.7 It is partially due to this body of knowledge that an ex vivo, long-term model of the intestinal epithelium started from native, non-transformed primary cells could be developed. Early attempts to produce such a culture system resulted in models in which the intestinal stem cells could transiently divide but would lose the ability to proliferate within weeks.8,9
The first published report of a culture system that could promote long-term survival of intestinal stem cells came from the group of Dr. Calvin Kuo.10 Through the culture of intestinal fragments containing epithelial and mesenchymal cells from neonatal mice, Ootani et al. devised an air-liquid interface model that, when supplemented with fetal bovine serum, produced cyst-like structures containing all major cells types of the adult mouse intestine. If fed appropriately, these cultures could be maintained for over one year.10
In the same year, Dr. Hans Clevers’ group published an alternative technique for culturing intestinal crypts in which sorted single LGR5+ cells from the adult mouse intestine were used as the starting material for the ex vivo tissue culture model. In lieu of the air-liquid interface, this system mimicked an extracellular matrix by culturing the crypts in a semi-solid, laminin/collagen-rich Matrigel® hemisphere. A cell culture medium mimicking the stem cell niche of the crypt was used that, in conjunction with the semi-solid matrix, enabled long-term culture of these cells. Compared to the cystic structures described by Ootani et al.,10 these “organoids” contained a central lumen surrounded by “buds” that represent the intestinal crypts (Figure 1). These crypt-like domains are functionally similar to those of the adult intestine, as dividing LGR5+ stem cells are intercalated with Paneth cells and located at the crypt base. These organoids therefore represent a highly physiologically relevant phenotype. In the system developed by the Clevers’ group, the LGR5+ stem cells divide to generate a self-renewing stem cell population, as well as cells that terminally differentiate into enterocytes, enteroendocrine cells or goblet cells. The terminally differentiated cells are ultimately extruded into the lumen from the villus-like domain, mimicking the physiological turnover of the adult intestinal epithelium. The organoids cultured in this manner can also be maintained indefinitely with periodic cell dissociation and passaging (Figure 2).5 This advancement thus represented the establishment of a novel model system that incorporates many physiological properties of in vivo mouse models with the ease-of-culture and ability to manipulate specific variables in the cellular environment that are typical of in vitro cell culture models.
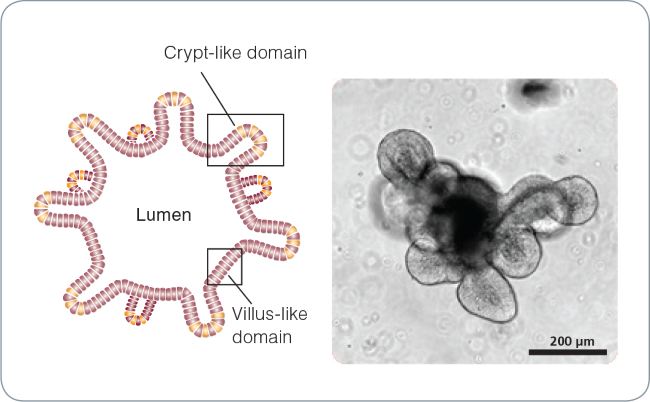
Figure 1. Intestinal organoid morphology.
Schematic of a mature intestinal organoid (left) and brightfield image of a mature (day 5) mouse intestinal organoid (right). The central lumen is surrounded by an epithelial monolayer with budding crypt-like domains.
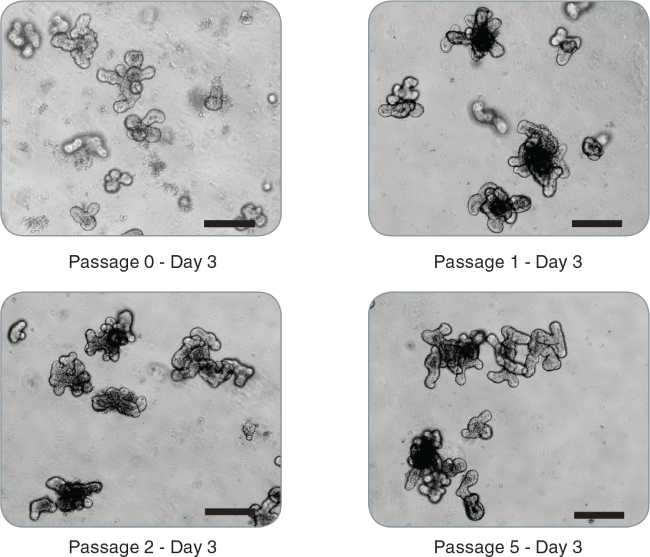
Figure 2. Passaging of intestinal organoids.
Representative images of mouse intestinal organoids from initial crypts: passage 0 - day 3, passage 1 - day 3, passage 2 - day 3, passage 5 - day 3 demonstrating maintenance of the budding phenotype (scale bar = 200 μm).
Culturing Intestinal Organoids
Development of a more consistent and stable cell culture medium that allows for the crypt-like structures of intestinal organoids to maintain their stemness has been a key factor in enabling successful culture of organoids. The intestinal stem cell niche requires a precise gradient of activity from multiple signaling pathways to maintain the proper homeostasis of the intestine. The cell culture medium published by Dr. Toshiro Sato from the Clevers’ group mimics this environment by including the cytokines EGF and R-Spondin1 to activate the EGF and WNT pathways, respectively, as well as Noggin to inhibit BMP signaling.5
Using Intestinal Organoids as a Model Culture System
Organoids are a valuable model for studying epithelial stem cell biology, as well as for structural and functional mechanisms of the intestine. Although this technology is still in its infancy, diverse proof-of-concept investigations have already been undertaken that validate the wide variety of applications. Organoids grown from mouse intestinal crypts can be subjected to changes in gene expression through transfection of DNA or small interfering RNA, as well as viral infection with adeno- or lentivirus.11 The rapid expansion of organoids in culture allows for their use in various analytical procedures including microarrays, sequencing, immunohistochemistry and mass spectrometry.12,13 Organoids can also be cryopreserved for long-term storage. Some recent studies that made use of organoid cultures can be grouped in the following categories of research:
Stem Cell Biology
Shortly before the development of organoid cultures, cell labeling and clonal cell analysis led to a more definitive understanding of stem cell dynamics within the intestinal crypt. It was known that an LGR5+ population at the crypt base undergoes stochastic symmetric divisions that maintain the stem cell pool and the transit amplifying population.6,14 Follow-up studies using organoids have identified a complex role for the intercalating Paneth cells in stem cell maintenance. Depletion of Paneth cells corresponds with a loss of LGR5+ cells from the crypt base.15 The Paneth cells have been shown to secrete cytokines, such as WNT, that are necessary for the maintenance of the stem cell niche. These cells have also been demonstrated to exhibit differential sensitivity to bacterial ligands, cytokines, and muscarinergic stimulation.15,16 As a clearer understanding of the specific roles of the individual cell types is established, it may be possible to target these cells for treatment of diseases arising from their abnormalities. An example of such targeting could be therapies directed at Paneth cell mutations in inflammatory bowel disease.
Human Disease
The organoid culture system incorporates key attributes that make it a valuable tool for studying human disease: first, organoids have been generated from intestinal stem cells isolated from human patient biopsies;17,18 and second, single sorted EPHB2+ cells, which reside at the crypt base, can grow into organoid cultures.18 Such organoids can be used to study the cellular mechanisms behind intestinal diseases through studies of gene expression or manipulation.
Inhibition of the mechanisms controlling proliferation and cell death play a role in many intestinal diseases. Disorders such as Crohn’s disease, ulcerative colitis, coeliac disease and sepsis all result in uncontrolled activation of the immune system, ultimately leading to intestinal damage. Microscopy and colorimetric assays using intestinal organoids have been designed to address gene function in the survival and cell death of epithelial cells.19
Organoids also provide a valuable tool for addressing the mechanisms of intestinal cancer ex vivo. Mouse intestinal organoids deficient in the adenomatous polyposis coli gene (Apc) display constitutively active WNT signaling and are tumorigenic when injected into nude mice, yielding highly proliferative tubular epithelial glands accompanied by prominent stromal tissue.15 Further studies were able to dissect the interactions between the MAPK and WNT signaling pathways in tumor progression. The oncogene BRAF is the most prevalent mutation in the sessile serrated adenomas and its overexpression in transgenic mice leads to upregulation of MAPK activity. Organoids derived from mice carrying the BRAFV600K mutation demonstrate a rapid development of generalized serrated dysplasia and also exhibit depletion of the stem cell pool at the crypt domain bases. This loss of stem cells was found to be antagonized by WNT/Beta- Catenin activity, demonstrating a possible fail-safe mechanism in which MAPK/BRAF signaling protects the intestinal tissue against oncogenic activation.20
The architecture of the organoid model also lends itself to studies specific to the immune system. Wilson et al. addressed the Paneth cell antimicrobial response by establishing an enteric infection model incorporating bacterial pathogen injection directly into the lumen of the organoids.21 This technical development offers a novel model to investigate host-microbe interactions.
Gene Therapy
Organoids can be subjected to viral infection with specific vectors. Organoids can also be transplanted into recipient mice after intestinal damage has been induced.22 These two attributes make it possible to address questions involving the correction of specific disease-causing mutations. Because organoids can be grown from single stem cells, clonal organoids can be generated and analyzed after transfection. This allows for precise transplantation of organoids with proper integration of introduced genetic material and makes a strong case for organoids as a cell-based therapeutic tool. Two recent studies focused on assay development and subsequent gene replacement therapy of the Cystic Fibrosis Transmembrane Conductance Receptor (CFTR) using organoid cultures as integral components. Dekkers et al. developed an organoid-based assay in which addition of the drug forskolin conveyed a cAMP-induced rapid swelling of wild-type organoids from both mouse and human intestinal samples. They demonstrated that the drug-induced swelling was greatly reduced in mice carrying the F508del mutation in CFTR, thus establishing a methodology to evaluate gene therapies targeting the CFTR mutation.23 Schwank et al. used this assay and made use of the CRISPR/CAS9 system in which, through induced homologous recombination, they could specifically correct the CFTR F508del allele. Organoids carrying the rescued allele regained the ability to swell when subjected to forskolin, demonstrating a proof-ofprinciple for gene replacement therapy.24
Other Applications
Beyond studies looking at specific signaling pathways, organoids also offer an opportunity for large-scale drug screening. Organoid-based screening can be either complementary to 2-dimensional screens performed using CACO-2 cells, or serve to replace these cell line-based assays with a more physiologically relevant model system.25 Proof-of-principle experiments using organoids to investigate the effect of small molecules on stem cell physiology have been performed. For example, treatment of organoids with a combination of valproic acid and small molecules inhibiting GSK3Beta and histone deacetylase resulted in a vast increase in LGR5+ stem cells and a 100-fold increase in colony-forming efficiency, suggesting a role of these molecules in stem cell maintenance.26 Further primary screening for effects of small molecules on wild-type and diseased organoids may prove to be a predictive tool to augment current ADMET (adsorption, distribution, metabolism, excretion, toxicology) methodologies. Spence et al. demonstrated that organoid derivation is not limited to stem cell isolation from primary tissue sources. This group demonstrated the ability to direct differentiation of human pluripotent stem cells into intestinal organoids in vitro using a temporal series of growth factor manipulations that mimic signals present during embryonic intestinal development. These human intestinal organoids differ from those cultured directly from intestinal tissue as they contain a mesenchymal layer that develops along with the epithelium in a manner similar to that observed during embryonic development.27
The ability to grow organoids that are genetically identical to individual patients represents a monumental step toward the elusive promise of individual patient disease diagnoses. Organoid cultures can be generated from normal and tumorous human intestines, a feature that makes it conceptually possible to use these cultures to screen for specific tumor-associated mutations and formulate more personalized treatment options. The HUB Foundation for Organoid Technology (http://hub4organoids.eu) is an example of an organization attempting to generate a human organoid library, or “biobank”, composed of matched healthy and diseased patient biopsies that can be subjected to multiple screening methods.
Since the first report that intestinal stem cells could be cultured ex vivo, a multitude of publications have been published that demonstrate the future utility of this technology in a number of fields, some of which have been reviewed here. A key factor to the early success of this novel culture method is how closely it recapitulates the structural and functional aspects of the mammalian intestinal epithelium. One of the most exciting aspects of organoid technology is the ability to adapt this organotypic culture method to other tissue types. To date, organoid cultures have been established from tissues originating from the liver,28 pancreas,29 Barrett’s esophagus,17 small intestine,17,18 colon,17,18 brain,30 lung31 and prostate,32,33 as well as from human induced pluripotent stem cells.27,34,35,36 The ability to replicate known functions of these tissues while exploring novel translational aspects such as transplantation make organoids an extremely exciting technology.
References
- van der Flier LG and Clevers H. Annu Rev Physiol 71: 241-260, 2009
- Barker N, et al. Nature 449: 1003-1007, 2007
- Buczacki SJA, et al. Nature 495: 65-69, 2013
- Barker N, et al. Genes Dev 22: 1856-1864, 2008
- Sato T, et al. Nature 459: 262-265, 2009
- Snippert HJ, et al. Cell 134-144, 2010
- Sambuy Y, et al. Cell Biol Toxicol 21: 1-26, 2005
- Evans GS, et al. J Cell Sci 101: 219-231, 1992
- Fukamachi H. J Cell Sci 103: 511-519, 1992
- Ootani A, et al. Nat Med 15: 701-706, 2009
- Koo B-K, et al. Nat Methods 9: 81-83, 2012
- Sato T and Clevers H. Science 340: 1190-1194, 2013
- Leushacke M and Barker N. Gut 63: 1345-1354, 2014
- Baker A-M, et al. Cell Rep 8: 940-947, 2014
- Sato T, et al. Nature 469: 415-418, 2011
- Farin HF, et al. J Exp Med 211: 1393-1405, 2014
- Sato T, et al. Gastroenterology 141: 1762-1772, 2011
- Jung P, et al. Nat Med 17: 1225-1227, 2011
- Grabinger T, et al. Cell Death Dis 5: e1228, 2014
- Riemer P, et al. Oncogene, 2014
- Wilson SS, et al. Mucosal Immunol, 2014
- Yui S, et al. Nat Med 18: 618-623, 2012
- Dekkers JF, et al. Nat Med 19: 939-945, 2013
- Schwank G, et al. Cell Stem Cell 13: 653-658, 2013
- Ranga A, et al. Adv Drug Deliv Rev 69-70: 19-28, 2014
- Yin X, et al. Nat Meth 11: 106-112, 2014
- Spence JR, et al. Nature 470: 105-109, 2011
- Huch M, et al. Nature 494: 247-250, 2013
- Huch M, et al. EMBO J 32: 2708-2721, 2013
- Lancaster MA, et al. Nature 501: 373-379, 2013
- Chapman HA, et al. J Clinic Invest 121: 2855-2862, 2011
- Gao D, et al. Cell 159: 176-187, 2014
- Karthaus WR, et al. Cell 159: 163-17520, 2014
- McCracken KW, et al. Nat Protoc. 6(12): 1920-28, 2011
- Watson CL, et al. Nat Med. 20(11): 1310-14, 2014
- McCracken KW, et al. Nature [Epub ahead of print], 2014
