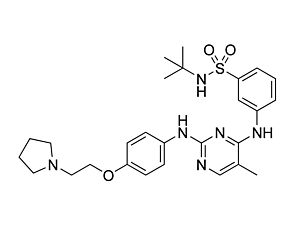
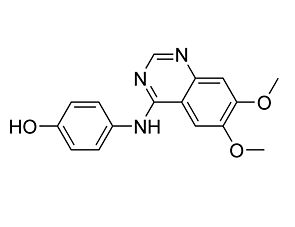
Ruxolitinib
JAK/STAT pathway inhibitor; Inhibits JAK1 and JAK2
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (3)
Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition.
Nature medicine 2014 SEP
Abstract
Alopecia areata (AA) is a common autoimmune disease resulting from damage of the hair follicle by T cells. The immune pathways required for autoreactive T cell activation in AA are not defined limiting clinical development of rational targeted therapies. Genome-wide association studies (GWAS) implicated ligands for the NKG2D receptor (product of the KLRK1 gene) in disease pathogenesis. Here, we show that cytotoxic CD8(+)NKG2D(+) T cells are both necessary and sufficient for the induction of AA in mouse models of disease. Global transcriptional profiling of mouse and human AA skin revealed gene expression signatures indicative of cytotoxic T cell infiltration, an interferon-γ (IFN-γ) response and upregulation of several γ-chain (γc) cytokines known to promote the activation and survival of IFN-γ-producing CD8(+)NKG2D(+) effector T cells. Therapeutically, antibody-mediated blockade of IFN-γ, interleukin-2 (IL-2) or interleukin-15 receptor β (IL-15Rβ) prevented disease development, reducing the accumulation of CD8(+)NKG2D(+) T cells in the skin and the dermal IFN response in a mouse model of AA. Systemically administered pharmacological inhibitors of Janus kinase (JAK) family protein tyrosine kinases, downstream effectors of the IFN-γ and γc cytokine receptors, eliminated the IFN signature and prevented the development of AA, while topical administration promoted hair regrowth and reversed established disease. Notably, three patients treated with oral ruxolitinib, an inhibitor of JAK1 and JAK2, achieved near-complete hair regrowth within 5 months of treatment, suggesting the potential clinical utility of JAK inhibition in human AA.
Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms.
Blood 2010
Abstract
Constitutive JAK2 activation in hematopoietic cells by the JAK2V617F mutation recapitulates myeloproliferative neoplasm (MPN) phenotypes in mice, establishing JAK2 inhibition as a potential therapeutic strategy. Although most polycythemia vera patients carry the JAK2V617F mutation, half of those with essential thrombocythemia or primary myelofibrosis do not, suggesting alternative mechanisms for constitutive JAK-STAT signaling in MPNs. Most patients with primary myelofibrosis have elevated levels of JAK-dependent proinflammatory cytokines (eg, interleukin-6) consistent with our observation of JAK1 hyperactivation. Accordingly, we evaluated the effectiveness of selective JAK1/2 inhibition in experimental models relevant to MPNs and report on the effects of INCB018424, the first potent, selective, oral JAK1/JAK2 inhibitor to enter the clinic. INCB018424 inhibited interleukin-6 signaling (50% inhibitory concentration [IC(50)] = 281nM), and proliferation of JAK2V617F(+) Ba/F3 cells (IC(50) = 127nM). In primary cultures, INCB018424 preferentially suppressed erythroid progenitor colony formation from JAK2V617F(+) polycythemia vera patients (IC(50) = 67nM) versus healthy donors (IC(50) textgreater 400nM). In a mouse model of JAK2V617F(+) MPN, oral INCB018424 markedly reduced splenomegaly and circulating levels of inflammatory cytokines, and preferentially eliminated neoplastic cells, resulting in significantly prolonged survival without myelosuppressive or immunosuppressive effects. Preliminary clinical results support these preclinical data and establish INCB018424 as a promising oral agent for the treatment of MPNs.
Therapeutic potential of JAK2 inhibitors.
Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program 2009
Abstract
The discovery of an activating tyrosine kinase mutation JAK2V617F in myeloproliferative neoplasms (MPNs), polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF) has resulted in the development of JAK2 inhibitors, of which several are being evaluated in phase I/II clinical studies. It is important to recognize that because the V617F mutation is localized in a region outside the adenosine triphosphate (ATP)-binding pocket of JAK2 enzyme, ATP-competitive inhibitors of JAK2 kinase (like the current JAK2 inhibitors in the clinic) are not likely to discriminate between wild-type and mutant JAK2 enzymes. Therefore, JAK2 inhibitors, by virtue of their near equipotent activity against wild-type JAK2 that is important for normal hematopoiesis, may have adverse myelosuppression as an expected side effect, if administered at doses that aim to completely inhibit the mutant JAK2 enzyme. While they may prove to be effective in controlling hyperproliferation of hematopoietic cells in PV and ET, they may not be able to eliminate mutant clones. On the other hand, JAK inhibitors may have great therapeutic benefit by controlling the disease for patients with MPNs who suffer from debilitating signs (eg, splenomegaly) or constitutional symptoms (which presumably result from high levels of circulating cytokines that signal through JAK enzymes). Indeed, the primary clinical benefits observed so far in MF patients have been significant reduction is splenomegaly, elimination of debilitating disease-related symptoms, and weight gain. Most importantly, patients with and without the JAK2V617F mutation appear to benefit to the same extent. In this review we summarize current clinical experience with JAK2 inhibitors in MPNs.
Related Products
Related Products

Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
