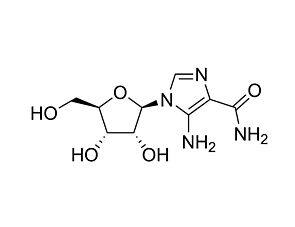
A769662
AMPK activator
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (7)
Role of AMP-activated protein kinase in regulating hypoxic survival and proliferation of mesenchymal stem cells.
Cardiovascular research 2014
Abstract
AIMS: Mesenchymal stem cells (MSCs) are widely used for cell therapy, particularly for the treatment of ischaemic heart disease. Mechanisms underlying control of their metabolism and proliferation capacity, critical elements for their survival and differentiation, have not been fully characterized. AMP-activated protein kinase (AMPK) is a key regulator known to metabolically protect cardiomyocytes against ischaemic injuries and, more generally, to inhibit cell proliferation. We hypothesized that AMPK plays a role in control of MSC metabolism and proliferation. METHODS AND RESULTS: MSCs isolated from murine bone marrow exclusively expressed the AMPKα1 catalytic subunit. In contrast to cardiomyocytes, a chronic exposure of MSCs to hypoxia failed to induce cell death despite the absence of AMPK activation. This hypoxic tolerance was the consequence of a preference of MSC towards glycolytic metabolism independently of oxygen availability and AMPK signalling. On the other hand, A-769662, a well-characterized AMPK activator, was able to induce a robust and sustained AMPK activation. We showed that A-769662-induced AMPK activation inhibited MSC proliferation. Proliferation was not arrested in MSCs derived from AMPKα1-knockout mice, providing genetic evidence that AMPK is essential for this process. Among AMPK downstream targets proposed to regulate cell proliferation, we showed that neither the p70 ribosomal S6 protein kinase/eukaryotic elongation factor 2-dependent protein synthesis pathway nor p21 was involved, whereas p27 expression was increased by A-769662. Silencing p27 expression partially prevented the A-769662-dependent inhibition of MSC proliferation. CONCLUSION: MSCs resist hypoxia independently of AMPK whereas chronic AMPK activation inhibits MSC proliferation, p27 being involved in this regulation.
Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells.
Cell cycle (Georgetown, Tex.) 2012 MAR
Abstract
The ability of somatic cells to reprogram their ATP-generating machinery into a Warburg-like glycolytic metabotype while overexpressing stemness genes facilitates their conversion into either induced pluripotent stem cells (iPSCs) or tumor-propagating cells. AMP-activated protein kinase (AMPK) is a metabolic master switch that senses and decodes intracellular changes in energy status; thus, we have evaluated the impact of AMPK activation in regulating the generation of iPSCs from nonstem cells of somatic origin. The indirect and direct activation of AMPK with the antidiabetic biguanide metformin and the thienopyridone A-769662, respectively, impeded the reprogramming of mouse embryonic and human diploid fibroblasts into iPSCs. The AMPK activators established a metabolic barrier to reprogramming that could not be bypassed, even through p53 deficiency, a fundamental mechanism to greatly improve the efficiency of stem-cell production. Treatment with metformin or A-769662 before the generation of iPSC colonies was sufficient to drastically decrease iPSC generation, suggesting that AMPK activation impedes early stem cell genetic reprogramming. Monitoring the transcriptional activation status of each individual reprogramming factor (i.e., Oct4, Sox2, Klf4 and c-Myc) revealed that AMPK activation notably prevented the transcriptional activation of Oct4, the master regulator of the pluripotent state. AMPK activation appears to impose a normalized metabolic flow away from the required pro-immortalizing glycolysis that fuels the induction of stemness and pluripotency, endowing somatic cells with an energetic infrastructure that is protected against reprogramming. AMPK-activating anti-reprogramming strategies may provide a roadmap for the generation of novel cancer therapies that metabolically target tumor-propagating cells.
AMP-activated protein kinase activator A-769662 is an inhibitor of the Na(+)-K(+)-ATPase.
American journal of physiology. Cell physiology 2009
Abstract
Muscle contraction and metabolic stress are potent activators of AMP-activated protein kinase (AMPK). AMPK restores energy balance by activating processes that produce energy while inhibiting those that consume energy. The role of AMPK in the regulation of active ion transport is unclear. Our aim was to determine the effect of the AMPK activator A-769662 on Na(+)-K(+)-ATPase function in skeletal muscle cells. Short-term incubation of differentiated rat L6 myotubes with 100 microM A-769662 increased AMPK and acetyl-CoA carboxylase (ACC) phosphorylation in parallel with decreased Na(+)-K(+)-ATPase alpha(1)-subunit abundance at the plasma membrane and ouabain-sensitive (86)Rb(+) uptake. Notably, the effect of A-769662 on Na(+)-K(+)-ATPase was similar in muscle cells that do not express AMPK alpha(1)- and alpha(2)-catalytic subunits. A-769662 directly inhibits the alpha(1)-isoform of the Na(+)-K(+)-ATPase, purified from rat and human kidney cells in vitro with IC(50) 57 microM and 220 microM, respectively. Inhibition of the Na(+)-K(+)-ATPase by 100 microM ouabain decreases sodium pump activity and cell surface abundance, similar to the effect of A-769662, without affecting AMPK and ACC phosphorylation. In conclusion, the AMPK activator A-769662 inhibits Na(+)-K(+)-ATPase activity and decreases the sodium pump cell surface abundance in L6 skeletal muscle cells. The effect of A-769662 on sodium pump is due to direct inhibition of the Na(+)-K(+)-ATPase activity, rather than AMPK activation. This AMPK-independent effect on Na(+)-K(+)-ATPase calls into question the use of A-769662 as a specific AMPK activator for metabolic studies.
Related Products
Related Products


Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
