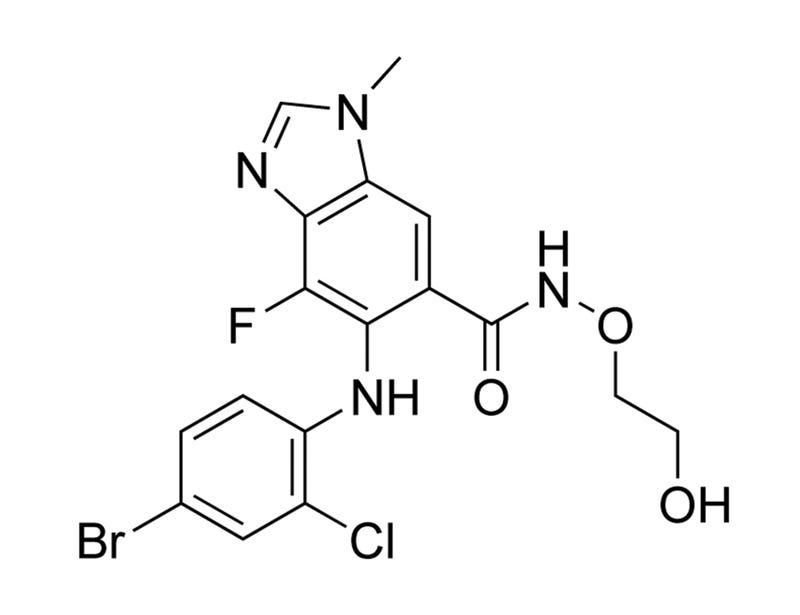
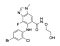
AZD6244
MEK/ERK pathway inhibitor; Inhibits MEK1 and MEK2
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Resources and Publications
Educational Materials (2)


Publications (6)
Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial.
JAMA 2014
Abstract
IMPORTANCE: Uveal melanoma is characterized by mutations in GNAQ and GNA11, resulting in mitogen-activated protein kinase pathway activation. OBJECTIVE: To assess the efficacy of selumetinib, a selective, non-adenosine triphosphate competitive inhibitor of MEK1 and MEK2, in uveal melanoma. DESIGN, SETTING, AND PARTICIPANTS: Randomized, open-label, phase 2 clinical trial comparing selumetinib vs chemotherapy conducted from August 2010 through December 2013 among 120 patients with metastatic uveal melanoma at 15 academic oncology centers in the United States and Canada. INTERVENTIONS: One hundred one patients were randomized in a 1:1 ratio to receive selumetinib, 75 mg orally twice daily on a continual basis (n = 50), or chemotherapy (temozolomide, 150 mg/m2 orally daily for 5 of every 28 days, or dacarbazine, 1000 mg/m2 intravenously every 21 days [investigator choice]; n = 51) until disease progression, death, intolerable adverse effects, or withdrawal of consent. After primary outcome analysis, 19 patients were registered and 18 treated with selumetinib without randomization to complete the planned 120-patient enrollment. Patients in the chemotherapy group could receive selumetinib at the time of radiographic progression. MAIN OUTCOMES AND MEASURES: Progression-free survival, the primary end point, was assessed as of April 22, 2013. Additional end points, including overall survival, response rate, and safety/toxicity, were assessed as of December 31, 2013. RESULTS: Median progression-free survival among patients randomized to chemotherapy was 7 weeks (95% CI, 4.3-8.4 weeks; median treatment duration, 8 weeks; interquartile range [IQR], 4.3-16 weeks) and among those randomized to selumetinib was 15.9 weeks (95% CI, 8.4-21.1 weeks; median treatment duration, 16.1 weeks; IQR, 8.1-25.3 weeks) (hazard ratio, 0.46; 95% CI, 0.30-0.71; P textless .001). Median overall survival time was 9.1 months (95% CI, 6.1-11.1 months) with chemotherapy and 11.8 months (95% CI, 9.8-15.7 months) with selumetinib (hazard ratio, 0.66; 95% CI, 0.41-1.06; P = .09). No objective responses were observed with chemotherapy. Forty-nine percent of patients treated with selumetinib achieved tumor regression, with 14% achieving an objective radiographic response to therapy. Treatment-related adverse events were observed in 97% of patients treated with selumetinib, with 37% requiring at least 1 dose reduction. CONCLUSIONS AND RELEVANCE: In this hypothesis-generating study of patients with advanced uveal melanoma, selumetinib compared with chemotherapy resulted in a modestly improved progression-free survival and response rate; however, no improvement in overall survival was observed. Improvement in clinical outcomes was accompanied by a high rate of adverse events. TRIAL REGISTRATION: clinicaltrials.gov Identifier: NCT01143402.
Comprehensive analysis of kinase inhibitor selectivity.
Nature biotechnology 2011
Abstract
We tested the interaction of 72 kinase inhibitors with 442 kinases covering textgreater80% of the human catalytic protein kinome. Our data show that, as a class, type II inhibitors are more selective than type I inhibitors, but that there are important exceptions to this trend. The data further illustrate that selective inhibitors have been developed against the majority of kinases targeted by the compounds tested. Analysis of the interaction patterns reveals a class of 'group-selective' inhibitors broadly active against a single subfamily of kinases, but selective outside that subfamily. The data set suggests compounds to use as tools to study kinases for which no dedicated inhibitors exist. It also provides a foundation for further exploring kinase inhibitor biology and toxicity, as well as for studying the structural basis of the observed interaction patterns. Our findings will help to realize the direct enabling potential of genomics for drug development and basic research about cellular signaling.
Identification of common predictive markers of in vitro response to the Mek inhibitor selumetinib (AZD6244; ARRY-142886) in human breast cancer and non-small cell lung cancer cell lines.
Molecular cancer therapeutics 2010
Abstract
Selumetinib (AZD6244; ARRY-142886) is a tight-binding, uncompetitive inhibitor of mitogen-activated protein kinase kinases (MEK) 1 and 2 currently in clinical development. We evaluated the effects of selumetinib in 31 human breast cancer cell lines and 43 human non-small cell lung cancer (NSCLC) cell lines to identify characteristics correlating with in vitro sensitivity to MEK inhibition. IC(50) textless1 micromol/L (considered sensitive) was seen in 5 of 31 breast cancer cell lines and 15 of 43 NSCLC cell lines, with a correlation between sensitivity and raf mutations in breast cancer cell lines (P = 0.022) and ras mutations in NSCLC cell lines (P = 0.045). Evaluation of 27 of the NSCLC cell lines with Western blots showed no clear association between MEK and phosphoinositide 3-kinase pathway activation and sensitivity to MEK inhibition. Baseline gene expression profiles were generated for each cell line using Agilent gene expression arrays to identify additional predictive markers. Genes associated with differential sensitivity to selumetinib were seen in both histologies, including a small number of genes in which differential expression was common to both histologies. In total, these results suggest that clinical trials of selumetinib in breast cancer and NSCLC might select patients whose tumors harbor raf and ras mutations, respectively.
Related Products
Related Products
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
