Tumorsphere Culture of Human Breast Cancer Cell Lines
.
- Document # 29936
- Version 1.1.0
- Jan 2014
Introduction
The idea of cancer stem cells was first introduced about forty years ago, when it was shown that leukemia cells had distinct sub-populations with varying proliferation rates. Since that time, the cancer stem cell field has grown and two cancer stem cell models have emerged. One model hypothesizes that a specific subset of cells within a tumor population is tumorigenic (i.e. cancer stem cells). The other model hypothesizes that due to the heterogeneity of cells and their activities found in cancer, only some cells are inherently tumorigenic and may exhibit stem cell-like qualities.1
Clarke et al. reported the existence of breast cancer stem cells which can be isolated using cell surface markers and functionally assessed in vivo by demonstrating de novo tumor formation.2 In addition, it has been demonstrated that primary human breast stem/progenitor cells can be enriched in non-adherent mammospheres,3 a concept adopted from the culture of neural stem cells as neurospheres.4 Recently, it was shown that human breast cancer cell lines were also capable of generating non-adherent spheres termed tumorspheres.5,6 Of these, certain breast cancer cell lines contain a sub-population with stem cell-like properties that are also enriched in tumorspheres and,5,6 because primary human breast cancer tissues are a challenge to obtain and even to culture, they are commonly used as a tool to evaluate breast cancer stem cell functionalities. This technical bulletin describes a method to culture breast cancer cell lines as tumorspheres using MammoCult™ medium from STEMCELL Technologies.
Protocol: Tumorsphere Culture of Breast Cancer Cell Lines*
*MDA-MB-231, MCF7, SKBR3, AU565, BT474, SUM149.Prior to tumorsphere culture, breast cancer cell lines are grown as monolayers according to the supplier’s recommendations on tissue culture-treated 10 cm dishes. Ensure that cells cultured as a monolayer are healthy and are 80 – 90% confluent. Over-grown cultures will have increased cell death.
- On Day 1 of the tumorsphere assay, remove one 80 – 90% confluent dish of cells from the incubator.
- Aspirate culture medium from the dish. Rinse twice with 5 mL of warm Hank’s Balanced Salt Solution (HBSS; Catalog #37250) to remove residual culture medium from the plate. Rinse
- Using a sterile cell scraper [BD Falcon, 1.8 cm blade; BD Catalog #353085], scrape cells gently from the dish. Note: Do not trypsinize cells at this stage as this could reduce tumorsphere forming efficiency.
- Resuspend scraped cells in 10 mL of complete MammoCult™ medium (Catalog #05620) and transfer to a 15 mL conical tube with a pipette. For detailed instructions on how to prepare complete MammoCult™ medium, please refer to the product information sheet.
- Centrifuge the cells at 500 x g for 3 minutes at room temperature (15°C – 25°C).
- Remove the tube from the centrifuge carefully, ensuring that the cell pellet is not disturbed. Carefully aspirate and discard the supernatant with a pipette.
- Resuspend the cell pellet into a single cell suspension with a 5 mL pipette in 2 mL of complete MammoCult™ medium.
- Prepare a dilution of cells to enumerate cell concentration. Please refer to the cell counting protocol for detailed instructions on enumerating cells with Trypan Blue (Catalog #07050).
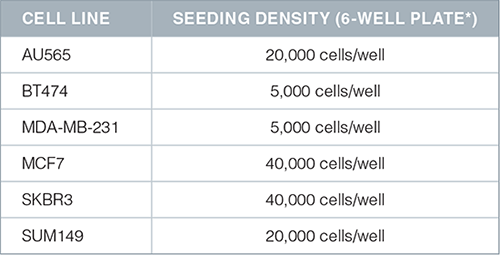
- Resuspend a pre-determined amount of cells in 2 mL of complete MammoCult™ medium (in triplicate) in each well of a 6 well ultra-low adherent plate (Catalog #27145). The seeding density for cell lines is typically in the range of 5,000 – 20,000 cells per well of a 6 well plate. Note: We recommend optimizing the seeding density for each cell line of interest.
- Recommended seeding densities for commonly used cell lines are shown below:
*Ultra-low adherent culture plates
- Incubate cultures in a 5% CO2, humidified incubator at 37°C for 7 days.
- Count the number of tumorspheres that are 60 μm or larger in size. The shape and appearance of the spheres is cell line-dependent. For example, tumorspheres will appear solid and will form tightly packed, round spheres (SUM149, BT474, MCF7) or looser and less rounded spheres (MDA-MB-231, AU565, SKBR3). See Figure 1A and Figure 2A.
Note: To passage spheres, refer to the tumorsphere passaging protocol
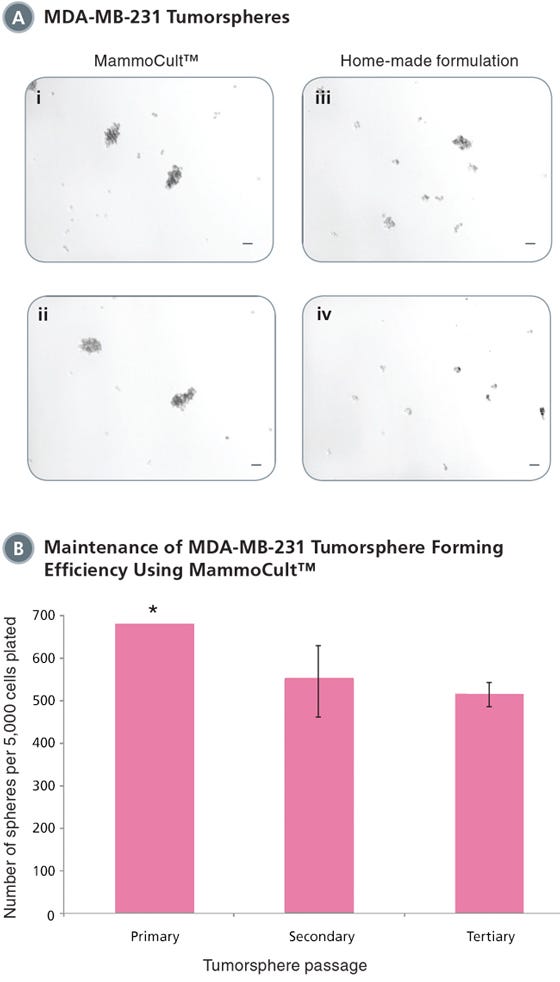
Figure 1. MammoCult™ supports the formation and maintenance of MDA-MB-231 tumorspheres in culture.
Each well was seeded with 5,000 cells per well and cultured for 7 days before imaging and counting. MDA-MB-231 is an invasive breast cancer cell line and will form more loose and less rounded tumorspheres. (A) MammoCult™ supports sphere formation of MDA-MB-231 cells better (i,ii) than a widely used home-made formulation (iii,iv). (B) Tumorspheres are established with high efficiency and can be passaged to secondary and tertiary tumorspheres in MammoCult™ medium without significant loss of the stem cell population.
*Sphere number estimated as there were greater than 500 spheres per field of view. Scale bar = 140 μm. Tumorsphere photos and data courtesy of Dr. S. Dunn and J. Law, Child & Family Research Institute (CFRI), Vancouver, Canada.
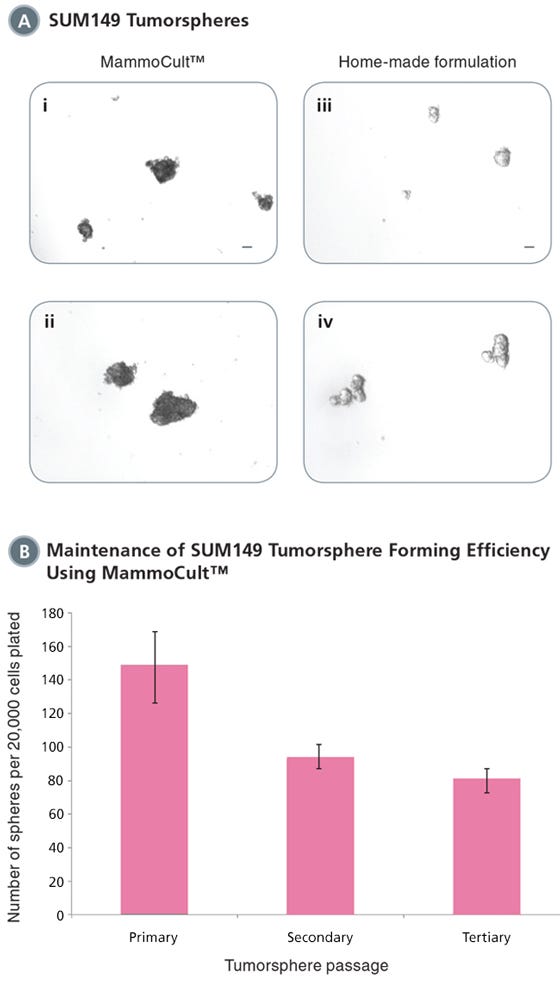
Figure 2. MammoCult™ supports the formation and maintenance of SUM149 tumorspheres in culture.
Each well was seeded with 20,000 cells per well and cultured for 7 days before imaging and counting. (A) SUM149 cells form tight mammospheres more efficiently in MammoCult™ (i,ii) than in the widely used home-made formulation (iii,iv). (B) SUM149 tumorspheres are established with high efficiency in MammoCult™. Tumorsphere forming efficiency remains high through secondary and tertiary passages.
Scale bar = 140 μm. Tumorsphere photos and data courtesy of Dr. S. Dunn and J. Law, Child & Family Research Institute (CFRI), Vancouver, Canada.
Protocol: Passaging Tumorspheres
- Harvest tumorspheres after 7 days in culture. Collect the entire culture into a 50 mL conical tube and centrifuge at 350 x g for 5 minutes.
Note: Tumorspheres should be passaged when they are ~60 μm in diameter and before they develop a dark center. Depending on the number of spheres present in each well, it may be necessary to pool multiple wells to obtain maximum number of tumorspheres for subsequent passages. - Aspirate as much supernatant as possible without disturbing the pellet. Note: The pellet may be very loose.
- Add 0.5 – 1 mL of pre-warmed Trypsin-EDTA (Catalog #07901). Use a P1000 pipette with a sterile plastic tip and set the volume to slightly less than the approximate volume of the remaining medium (for example: if volume of remaining medium is 800 μL, set the volume of the pipettor to 700 μL to avoid creating bubbles). Pre-wet the tip with complete MammoCult™ medium to reduce cells sticking inside the tip. For detailed instructions on how to prepare complete MammoCult™ medium (Catalog #05620), please refer to the product information sheet.
- Triturate tumorspheres by slightly tilting the tip and pressing it against the bottom or side of the tube to generate resistance in order to break up the tumorspheres. Rinse the side of the tube during trituration to remove the remaining spheres that are attached to the side of the tube. If some tumorspheres are not dissociated after 1.5 minutes (this usually occurs at later passages), trituration can be extended to a maximum of 2 minutes.
- Add 5 mL of cold HBSS (Catalog #37250) + 2% FBS (Catalog #06100) and centrifuge the cell suspension at 350 x g for 5 minutes.
- Aspirate the supernatant and resuspend the pellet in 0.5 mL of complete MammoCult™ medium. Perform a viable cell count using Trypan Blue (Catalog #07050) according to the cell counting protocol.
Complete MammoCult™ medium can be used to generate tumorspheres that can be maintained over several passages. See Figures 1B and 2B.
Protocol: Cell Counting
Use a hemacytometer that has a 0.100 mm depth chamber (e.g. Neubauer hemacytometer).
Trypan Blue Exclusion
- Trypan Blue stains dead cells but does not stain live cells. The dye enters cells with compromised membranes to stain them blue. Thus, live cells can be distinguished from dead cells by their ability to exclude the blue dye. Ensure that the cell suspension to be counted is completely resuspended. Before the cells settle, place a suitable volume of a cell suspension (20-200 μL) in a centrifuge tube.
- Add an equal volume of 0.4% Trypan Blue (Catalog #07050) and gently mix. Incubate the mixture for 5 minutes at room temperature (15°C – 25°C). Resuspend the cell mixture after incubation.
- Prepare a hemacytometer by first cleaning the chamber surface with 70% ethanol. Wipe dry. Position the cover-slip over the chambers.
- Using a P20 pipette, place 10 μL of stained cells into the hemacytometer chamber.
- Using a hand tally counter, count the number of viable (unstained) cells in an area of 16 squares. When counting, always count only live (i.e. bright and not blue) cells.
- Move the hemacytometer to another set of 16 squares and carry on counting until all 4 sets of 16 corner squares are counted. See Figure 3.
- Total the cell count from all 4 sets of 16 corner squares. Divide this number by 4 to find the average number. Multiply the average number by 2 (to adjust for the 1:2 dilution factor with trypan blue). Then, multiply by 1 x 104 to obtain the concentration of cells per mL.
Example: If the total cell count from all 4 sets of squares is 350 and the dilution factor is 2, then the concentration of cells is:
350/4 x 2 x (1 x 104) = 1.75 x 106 cells per mL.
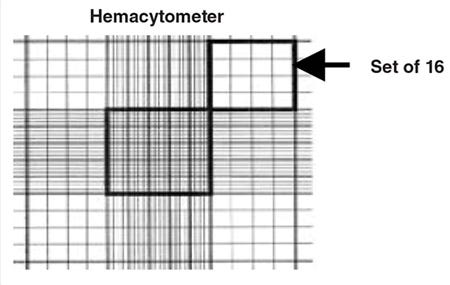
Figure 3. A sketch of the grid seen on a hemacytometer. Cells are counted in the four sets of 16 squares at each corner of the grid.
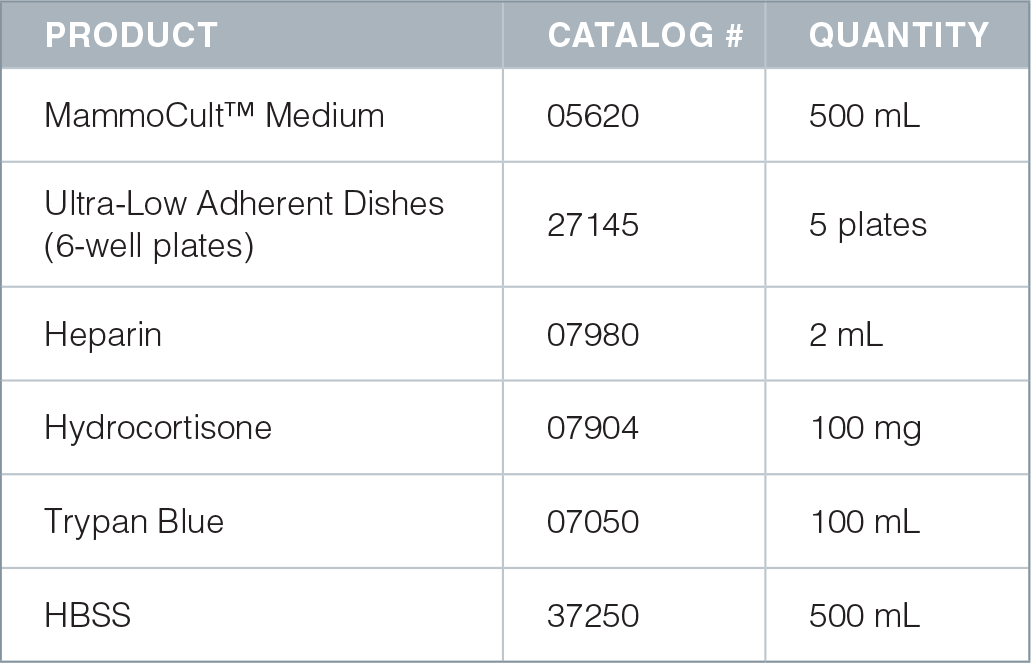
Product Information
References
- Charafe-Jauffret E, et al. Pathobiology. 75: 75 – 84, 2008
- Al-Hajj M, et al. PNAS. 100: 3983 – 3988, 2003
- Dontu G, et al. Genes and Dev. 17: 1253 – 1270, 2003
- Reynolds BA, et al. Science. 255: 1707 – 1710, 1992
- Fillmore CM, et al. Breast. Can Res. 2:R25. Epub 2008
- Charafe-Jauffret E, et al. Cancer Res. 69: 1302 – 1313, 2009
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration

