Thank you for your interest in this product.
Please provide us with your contact information and your local representative
will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
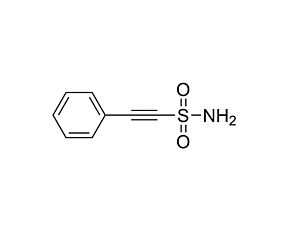
(±)-Nutlin-3 is a small-imidazoline-based mouse double minute 2 (MDM2) protein antagonist which disrupts MDM2-p53 interaction (IC₅₀ = 0.09 μM; Vassilev et al.). MDM2 binds the p53 tumor suppressor protein with high affinity and negatively modulates its transcriptional activity and stability. By disrupting this interaction, (±)-Nutlin-3 therefore promotes the expression of p53-regulated genes and exhibits potent antiproliferative activity in cells with functional p53, but not in cells with mutated p53 (Vassilev et al., Gu et al.).
CANCER RESEARCH
· Inhibits the proliferation of exponentially growing human skin fibroblasts (IC₅₀ = 2.2 μM) and mouse embryonic fibroblasts (IC₅₀ = 1.3 μM), and suppresses the growth of established tumor xenografts in mice (Vassilev et al.).
· Leads to G1 cell-cycle arrest in HCT116 colon carcinoma cell line expressing wild-type p53 and p21 (Benson et al.).
· Induces p53-mediated apoptosis in solid tumor and pediatric acute lymphoblastic leukemia cell lines (Gu et al.; Vaseva et al.)
Cell Type
Cancer Cells and Cell Lines, Leukemia/Lymphoma Cells, Mouse Embryonic Fibroblasts
p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes.
Benson EK et al.
Oncogene 2014 JUL
Abstract
The p53 tumor suppressor protein is a major sensor of cellular stresses, and upon stabilization, activates or represses many genes that control cell fate decisions. While the mechanism of p53-mediated transactivation is well established, several mechanisms have been proposed for p53-mediated repression. Here, we demonstrate that the cyclin-dependent kinase inhibitor p21 is both necessary and sufficient for the downregulation of known p53-repression targets, including survivin, CDC25C, and CDC25B in response to p53 induction. These same targets are similarly repressed in response to p16 overexpression, implicating the involvement of the shared downstream retinoblastoma (RB)-E2F pathway. We further show that in response to either p53 or p21 induction, E2F4 complexes are specifically recruited onto the promoters of these p53-repression targets. Moreover, abrogation of E2F4 recruitment via the inactivation of RB pocket proteins, but not by RB loss of function alone, prevents the repression of these genes. Finally, our results indicate that E2F4 promoter occupancy is globally associated with p53-repression targets, but not with p53 activation targets, implicating E2F4 complexes as effectors of p21-dependent p53-mediated repression.
Blockade of Hsp90 by 17AAG antagonizes MDMX and synergizes with Nutlin to induce p53-mediated apoptosis in solid tumors.
Vaseva AV et al.
Cell death & disease 2011 JAN
Abstract
Strategies to induce p53 activation in wtp53-retaining tumors carry high potential in cancer therapy. Nutlin, a potent highly selective MDM2 inhibitor, induces non-genotoxic p53 activation. Although Nutlin shows promise in promoting cell death in hematopoietic malignancies, a major roadblock is that most solid cancers do not undergo apoptosis but merely reversible growth arrest. p53 inhibition by unopposed MDMX is one major cause for apoptosis resistance to Nutlin. The Hsp90 chaperone is ubiquitously activated in cancer cells and supports oncogenic survival pathways, many of which antagonize p53. The Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17AAG) is known to induce p53-dependent apoptosis. We show here that in multiple difficult-to-kill solid tumor cells 17AAG modulates several critical components that synergize with Nutlin-activated p53 signaling to convert Nutlin's transient cytostatic response into a cytotoxic killing response in vitro and in xenografts. Combined with Nutlin, 17AAG destabilizes MDMX, reduces MDM2, induces PUMA and inhibits oncogenic survival pathways, such as PI3K/AKT, which counteract p53 signaling at multiple levels. Mechanistically, 17AAG interferes with the repressive MDMX-p53 axis by inducing robust MDMX degradation, thereby markedly increasing p53 transcription compared with Nutlin alone. To our knowledge Nutlin+17AAG represents the first effective pharmacologic knockdown of MDMX. Our study identifies 17AAG as a promising synthetic lethal partner for a more efficient Nutlin-based therapy.
MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2.
Gu L et al.
Leukemia 2008 APR
Abstract
In pediatric acute lymphoblastic leukemia (ALL), overexpression of murine double minute 2 (MDM2) protein by leukemic cells is typically associated with a wild-type (wt)-p53 phenotype and chemoresistance. A recently developed small-molecule antagonist of MDM2, nutlin-3, inhibits the MDM2-p53 interaction, resulting in induction of p53 activity and apoptosis. In this study, we evaluated the cytotoxic effect of nutlin-3 on ALL cells with different p53 status and MDM2 expression, using 18 cell lines and 30 primary leukemia samples. We found that both ALL cell lines and primary ALL samples with wt-p53 are sensitive to nutlin-3. No cytotoxic effect of nutlin-3 was detected in ALL cells with either p53-mutant or -null phenotype. In wt-p53 ALL cells, there was a significant positive correlation between MDM2 expression levels and sensitivity to nutlin-3. Nutlin-3-induced cell death was mediated by p53-induced activation of proapoptotic proteins and by p53-induced repression of the anti-apoptotic protein survivin. As p53 function is inhibited by MDM2 in chemoresistant, MDM2-overexpressing ALL cells, potent killing of these cells by nutlin-3 suggests that this agent may be a novel therapeutic for refractory ALL.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.