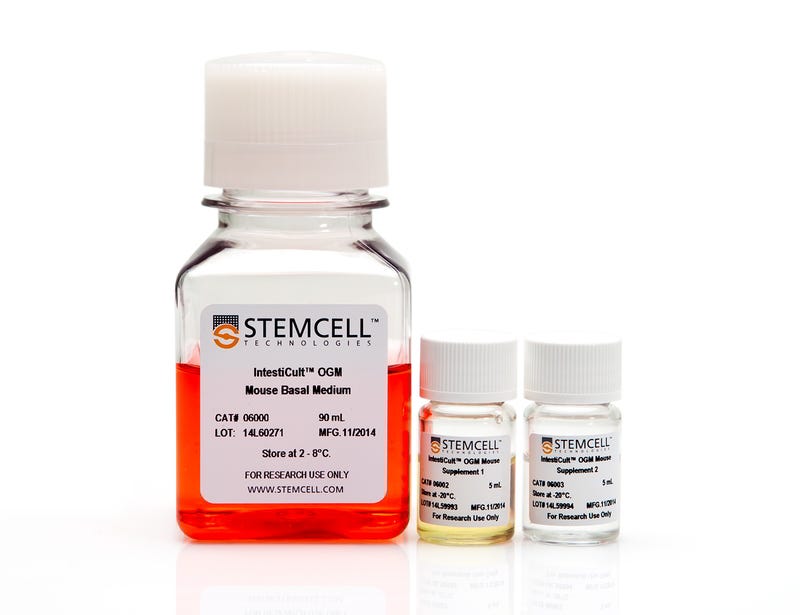
IntestiCult™ Organoid Growth Medium (Mouse)
Cell culture medium for establishment and maintenance of mouse intestinal organoids
Request Pricing
Thank you for your interest in this product. Please provide us with your contact information and your local representative will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
-
 Gentle Cell Dissociation Reagent
Gentle Cell Dissociation ReagentcGMP, enzyme-free cell dissociation reagent
-
 DMEM/F-12 with 15 mM HEPES
DMEM/F-12 with 15 mM HEPESDulbecco's Modified Eagle's Medium/Nutrient Ham's Mixture F-12 (DMEM/F-12) with 15 mM HEPES buffer
-
 D-PBS (Without Ca++ and Mg++)
D-PBS (Without Ca++ and Mg++)Dulbecco’s phosphate-buffered saline without calcium and magnesium
Overview
Protocols and Documentation
Find supporting information and directions for use in the Product Information Sheet or explore additional protocols below.
Applications
This product is designed for use in the following research area(s) as part of the highlighted workflow stage(s). Explore these workflows to learn more about the other products we offer to support each research area.
Resources and Publications
Educational Materials (39)







































Publications (95)
Abstract
Abstract
Abstract
Related Products

Legal Statement:
This product was developed under a license to intellectual property owned by Hubrecht Organoid Technology (HUB). This product is sold for research use only. Purchase of this product does not include the right to use it for drug screening aiming for commercial gain, equipment validation, biobanking, or for other commercial purposes. Purchasers wishing to use the product for purposes other than basic research use should contact HUB at www.huborganoids.nl to obtain a further license. Purchasers may apply for a License from HUB, which will not be unreasonably withheld by HUB.
Quality Statement:
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.
