Thank you for your interest in this product.
Please provide us with your contact information and your local representative
will contact you with a customized quote. Where appropriate, they can also assist you with a(n):
Estimated delivery time for your area
Product sample or exclusive offer
In-lab demonstration
By submitting this form, you are providing your consent to STEMCELL Technologies Canada Inc. and its subsidiaries and affiliates (“STEMCELL”) to collect and use your information, and send you newsletters and emails in accordance with our privacy policy. Please contact us with any questions that you may have. You can unsubscribe or change your email preferences at any time.
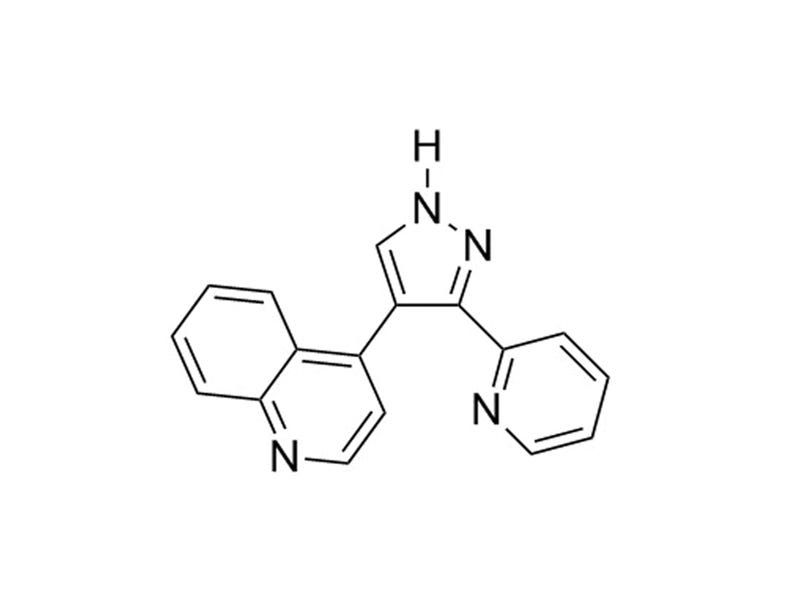
LY364947 is a selective inhibitor of the TGFβ/Activin/NODAL pathway that inhibits ALK5 (IC₅₀ = 59 nM)(Sawyer et al.). LY364947 less effectively inhibits TGFβRII (IC₅₀ = 400 nM), p38 MAPK (IC₅₀ = 740 nM), and mixed lineage kinase-7 (MLK-7; IC₅₀ = 1,400 nM) (Li et al., 2006; Sawyer et al.).
REPROGRAMMING
· In combination with valproic acid, can replace SOX2 in reprogramming of mouse embryonic fibroblasts transduced with OCT4, KLF4 and c-MYC (Ichida et al.).
DIFFERENTIATION
· Blocks chondrogenesis induced by mechanical load in human mesenchymal stem cells (Li et al., 2010).
· Restores the hematopoietic potential of mouse para-aortic splanchnopleural cells deficient for the Evi-1 transcription factor (Sato et al.).
· Impairs definitive endoderm differentiation competence in human embryonic stem (ES) cells (Jaremko et al.).
· Blocks TGF-β-induced endothelial-to-mesenchymal transition of NMuMg mammary epithelial cells or mouse ES cell-derived endothelial cells (Peng et al.; Kokudo et al.).
CANCER RESEARCH
· Suppresses colony-forming ability of mouse and human leukemia-initiating cells cultured with OP-9 stromal cells, and, when combined with imatinib, reduces lethality in a mouse model of chronic myeloid leukemia (Naka et al.).
· Reduces invasiveness of MDA-MB-231 breast cancer cells in a matrigel invasion assay (Shiou et al.).
Cell Type
Cancer Cells and Cell Lines, Chondrocytes, Endothelial Cells, Hematopoietic Stem and Progenitor Cells, Mammary Cells, Pluripotent Stem Cells
This product is designed for use in the following research area(s) as part
of the highlighted workflow stage(s). Explore these workflows to learn more about the other products we
offer to support each research area.
Regulation of developmental competence and commitment towards the definitive endoderm lineage in human embryonic stem cells.
Jaremko KL and Marikawa Y
Stem cell research 2013 MAY
Abstract
Human embryonic stem cells (hESCs) can self-renew and become all three germ layers. Nodal/Activin signaling specifies developmental status in hESCs: moderate Nodal/Activin signaling maintains pluripotency, while enhancement and inhibition promote definitive endoderm (DE) and neuroectoderm (NE) development, respectively. However, how modulation of Nodal/Activin signaling influences developmental competence and commitment toward specific lineages is still unclear. Here, we showed that enhancement of Nodal/Activin signaling for 4 days was necessary and sufficient to upregulate DE markers, while it diminished the upregulation of NE markers by inhibition of Nodal/Activin signaling. This suggests that after 4 days of enhanced Nodal/Activin signaling, hESCs are committed to the DE lineage and have lost competence toward the NE lineage. In contrast, inhibition of Nodal/Activin signaling using LY364947 for 2 days was sufficient to impair competence toward the DE lineage, although cells were still able to activate LEFTY1 and NODAL, direct targets of Nodal/Activin signaling. Expression analyses indicated that the levels of pluripotency regulators NANOG and POU5F1 were significantly diminished by 2 days of LY364947 treatment, although the expression of NANOG, but not POU5F1, was restored immediately upon Activin A treatment. Thus, downregulation of POU5F1 coincided with the abrogation of DE competence caused by inhibition of Nodal/Activin signaling.
Mechanical load modulates chondrogenesis of human mesenchymal stem cells through the TGF-beta pathway.
Li Z et al.
Journal of cellular and molecular medicine 2010 JUN
Abstract
This study investigated the effect of mechanical load on human mesenchymal stem cell (hMSC) differentiation under different exogenous transforming growth factor-beta1 (TGF-beta(1)) concentrations (0, 1 or 10 ng/ml).The role of the TGF-beta signalling pathway in this process was also studied. Human MSCs were seeded into fibrin-biodegradable polyurethane scaffolds at a cell density of 5 x 10(6) cells per scaffold and stimulated using our bioreactor. One hour of surface motion superimposed on cyclic compression was applied once a day over seven consecutive days. Scaffolds were analysed for gene expression, DNA content and glycosaminoglycan amount. Addition of TGF-beta(1) in the culture medium was sufficient to induce chondrogenesis of hMSCs. Depending on the TGF-beta(1) concentration of the culture medium, mechanical load stimulated chondrogenesis of hMSCs compared to the unloaded scaffolds, with a much stronger effect on gene expression at lower TGF-beta(1) concentrations. With TGF-beta(1) absent in the culture medium, mechanical load stimulated gene transcripts and protein synthesis of TGF-beta(1) and TGF-beta(3). TGF-beta type I receptor inhibitor LY364947 blocked the up-regulation on TGF-beta(1) and TGF-beta(3) production stimulated by mechanical load, and also blocked the chondrogenesis of hMSCs. Taken together, these findings suggest that mechanical load promotes chondrogenesis of hMSCs through TGF-beta pathway by up-regulating TGF-beta gene expression and protein synthesis.
TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia.
Naka K et al.
Nature 2010 FEB
Abstract
Chronic myeloid leukaemia (CML) is caused by a defined genetic abnormality that generates BCR-ABL, a constitutively active tyrosine kinase. It is widely believed that BCR-ABL activates Akt signalling that suppresses the forkhead O transcription factors (FOXO), supporting the proliferation or inhibiting the apoptosis of CML cells. Although the use of the tyrosine kinase inhibitor imatinib is a breakthrough for CML therapy, imatinib does not deplete the leukaemia-initiating cells (LICs) that drive the recurrence of CML. Here, using a syngeneic transplantation system and a CML-like myeloproliferative disease mouse model, we show that Foxo3a has an essential role in the maintenance of CML LICs. We find that cells with nuclear localization of Foxo3a and decreased Akt phosphorylation are enriched in the LIC population. Serial transplantation of LICs generated from Foxo3a(+/+) and Foxo3a(-/-) mice shows that the ability of LICs to cause disease is significantly decreased by Foxo3a deficiency. Furthermore, we find that TGF-beta is a critical regulator of Akt activation in LICs and controls Foxo3a localization. A combination of TGF-beta inhibition, Foxo3a deficiency and imatinib treatment led to efficient depletion of CML in vivo. Furthermore, the treatment of human CML LICs with a TGF-beta inhibitor impaired their colony-forming ability in vitro. Our results demonstrate a critical role for the TGF-beta-FOXO pathway in the maintenance of LICs, and strengthen our understanding of the mechanisms that specifically maintain CML LICs in vivo.
PRODUCTS ARE FOR RESEARCH USE ONLY AND NOT INTENDED FOR HUMAN OR ANIMAL DIAGNOSTIC OR THERAPEUTIC USES UNLESS OTHERWISE STATED. FOR ADDITIONAL INFORMATION ON QUALITY AT STEMCELL, REFER TO WWW.STEMCELL.COM/COMPLIANCE.